

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Epithelial ovarian cancer is the leading cause of disease-related deaths from gynecologic malignancies, and its incidence and mortality rates in Korea are increasing.12 In addition to well-known prognostic factors, such as stage, histology, grade, and residual disease after surgery,3 clinical studies are underway to identify potentially actionable mutations in ovarian cancer through a greater understanding of molecular mechanisms and to evaluate therapeutic agents of these mutations.45
Next generation sequencing (NGS) is able to reveal genomic aberrations by harnessing its massively parallel sequencing capability to analyze multiple genes simultaneously in a single assay. Moreover, this technology has recently become more affordable, leading to large collaborative studies on whole genomes that have been able to document targetable genes and predictive biomarkers in cancer.67 As of March 2017, the National Health Insurance system in Korea has paid the cost of NGS panels for several types of solid tumors, including ovarian cancer, and the number of NGS tests has increased exponentially.
We reviewed retrospective data of 84 patients who underwent NGS and reported our experiences with integrating an NGS panel into clinical practice in ovarian cancer. We identified potentially actionable genomic alterations and used them to evaluate the therapeutic utility of individual treatment options.
MATERIALS AND METHODS
Patient samples
Between March 1, 2017 and July 31, 2018, 84 tumor samples from ovarian cancer patients treated at Yonsei Cancer Center were subjected to NGS. The tumor samples were prepared from formalin-fixed, paraffin-embedded (FFPE) tissues. An expert pathologist (H.S.K.) reviewed hematoxylin and eosinstained slides to ensure that ≥20% of the nucleated cells in the sample were derived from the tumor. Tumor specimens were macrodissected after a hematoxylin-eosin reference slide check to ensure the proportion of tumor content. For DNA and RNA extraction, two to five slides of resected specimens of a thickness of 5 µm were needed. A board-certified gynecological pathologist diagnosed all cases. We performed a retrospective review of patient medical records, including age, histologic type, stage as defined by the International Federation of Gynecology and Obstetrics, and the timing of the NGS test.
NGS
We performed NGS analysis of 84 FFPE cancers with sufficiently high tumor cellularity (>30%). Genomic DNA was extracted using a Maxwell CSC DNA FFPE Kit (Promega, Madison, WI, USA), according to the manufacturer's instructions. The products were sequenced on a MiSeq System (Illumina, San Diego, CA, USA). Mutational and copy number analyses were performed using a TruSight Tumor 170 panel (Illumina) that covers, respectively, 170 genes and 59 genes for mutational and copy number analyses (Supplementary Table 1, only online). For mutational analysis, FASTQ files were uploaded on the Illumina BaseSpace software (Illumina) for variant interpretation. Only variants in coding regions and promoter regions or splice variants were retained. In addition, we retained only variants present in <1% of the population, according to ExAC and 1000 genomes, and also present in >5% of reads with a minimum read depth of 250. All retained variants were reviewed against reference websites [Catalogue of Somatic Mutations in Cancer (http://evs.gs.washington.edu/EVS/), Precision Oncology Knowledge Base (http://oncokb.org), and dbSNP (https://www.ncbi.nlm.nih.gov/snp)]. Only pathogenic variants were selected. In copy number analysis, only genes with a more than two-fold change relative to the average level were considered for amplification. We also performed total nucleic acid extraction to obtain ribonucleic acid (RNA). An Archer FusionPlex Solid Tumor Kit (ArcherDx, Boulder, CO, USA) was used to analyze the RNA for fusions and splice variants: the kit covers 55 genes.8 Specimens yielded more than 40 ng of DNA and RNA. DNA fragment sizes of at least 79 bp and RNA fragment sizes of at least 63 bp were selected for targeted sequencing. Our goal in this study was to assess the feasibility and utility of using the Illumina MiSeq platform to integrate a NGS panel into a real-world setting of ovarian cancer clinical practice.
Data interpretation
Actionable somatic alterations are defined as those that could be targeted by a drug available for on-label, off-label, or in clinical trials. These alterations were selected based on a literature search of the MD Anderson Knowledge Base for Precision Medicine (http://PCT.MDAnderson.org), The Cancer Genome Atlas (TCGA) (http://cancergenome.nih.gov/), and genes related to homologous recombination repair (HRR) (Supplementary Table 2, only online).
We used the following guidelines to classify these alterations into four tiers.9 Tier 1 comprised known tumor type-specific actionable somatic mutations of confirmed clinical utility in predicting responses to U.S. Food and Drug Administration (FDA)-approved therapies, prognoses, diagnoses, or increased risk of inherited cancer. Tier 2 included actionable somatic mutations in other tumor types or somatic mutations in targetable pathways with potential clinical significance, such as susceptibility to FDA-approved therapies, prognoses, diagnoses, or increased risk of inherited cancer. Alterations of unknown clinical significance were classified as Tier 3, and those considered benign or likely so were grouped in Tier 4.
Clinical implications
We defined the term clinical implication as the capability of NGS results to provide useful information about patients and their family members that could be used to diagnose, monitor, predict the occurrence of disease and to create informed choices about treatment options.10 These clinical implications were categorized into three categories to evaluate the clinical impact of NGS results: 1) those who received targeted therapy; 2) identification of potential candidates for targeted therapy; and 3) genetic counseling for the patient and other at-risk family members.
RESULTS
Patient clinicopathologic characteristics
A total of 227 ovarian cancer patients were treated in our institution between March 1, 2017 and July 31, 2018, and 84 (37%) patients underwent NGS analysis. Table 1 illustrates the baseline clinicopathological characteristics of the 84 patients who underwent NGS. The median age of this group was 54 years (range, 34–77). The most common histologic type was high-grade serous carcinoma (65.1%). Sixty-eight (80.7%) patients had advanced-stage disease (Stages III/IV). Forty-nine (57.8%) patients underwent primary debulking surgery (PDS), and 35 (42.2%) underwent neoadjuvant chemotherapy (NAC). Thirty-nine (45.8%) patients underwent NGS during PDS: 8 (9.6%) during pre-NAC, 26 (31.3%) during interval debulking surgery (IDS), and 11 (13.3%) patients at the time of relapse.
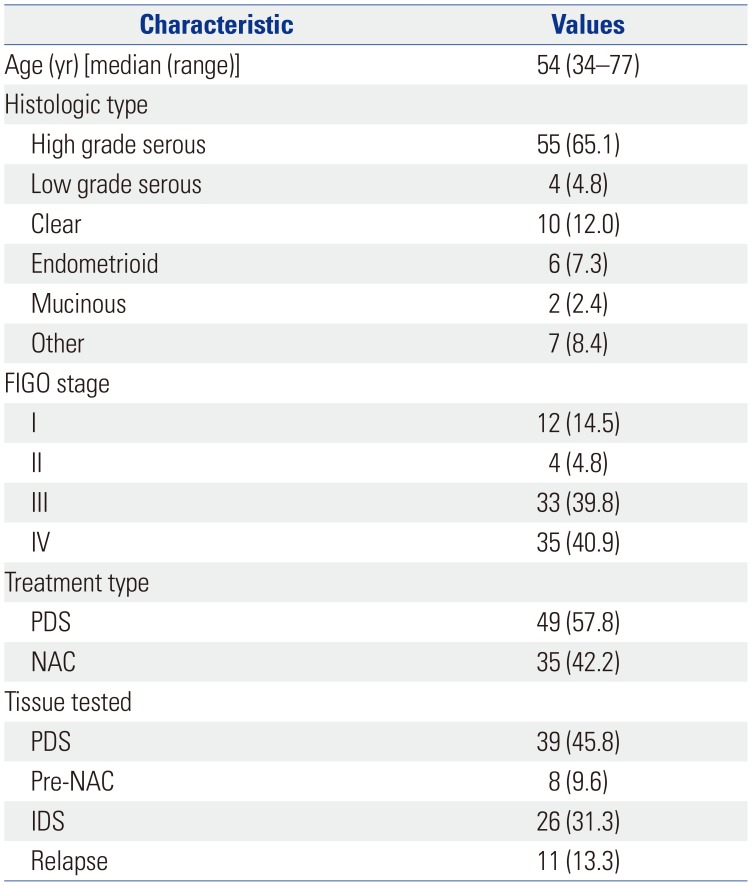
Table 1
Clinicopathological Characteristics (n=84)
![]()
Genomic alterations
All patients had at least one genomic alteration. The mean number of mutations per patient was 10.5. Fifty-seven (67.9%) patients had more than one actionable alteration other than TP53. Of the 57 patients, 16 (28.6%) had a mutation in HRR-related genes (Fig. 1). In addition, we analyzed the distribution of patients with somatic BRCA mutations at each time point of NGS analysis. Of the 11 patients with somatic BRCA mutations, there were 2 patients (2/8, 25.0%) in the Pre-NAC group, 5 patients (6/39, 15.4%) in the PDS group, 2 patients (2/26, 7.7%) in the IDS group, and 2 patients (1/11, 9.1%) in the relapse group. The chemo-naive group (pre-NAC, PDS) and the chemotherapy group (IDS, relapse) comprised 8/47 (17.0%) and 3/37 (8.1%), respectively. We also reviewed the Germline BRCA status for the patients (Supplementary Table 3, only online). Of the 84 patients, 12 (14.3%) had germline BRCA1/2 mutation, 50 (59.5%) had no germline BRCA1/2 mutation, and 26 (31.0%) did not undergo the germline BRCA test. Table 2 shows the tumor molecular profiles and clinical utility of actionable somatic mutations. Among single nucleotide variants and indel in tiers 1 or 2, the most frequently identified mutations were in TP53 (64%), PIK3CA (15%), and BRCA1/2 (17%) (Fig. 2A). Among copy number variations and fusions, the most frequently identified mutations were in MYC (27%), TFRC (24%), and CCNE1 (13%) (Fig. 2B). The most commonly mutated genes among the HRR-related genes in tiers 1 or 2 included BRCA1 (11%), BRCA2 (9%), ATM (4%), and CHEK1 (4%) (Fig. 2C). The most frequently mutated genes among the TCGA druggable genes in tiers 1 and 2 were KRAS (16%), CCNE1 (13%), ERBB2 (5%), RICTOR (1%), and ERBB3 (1%) (Fig. 2C). Identified mutations were categorized into six pathways or functional groups: cell cycle (RB1, CCNE1, CDK2, CCND1, CDK4, CDK6, CDKN2A, MYC, SRC, JAK1, JAK2, STAT1, STAT3), DNA damage response (CHEK1, CHEK2, BRCA1, BRCA2, MLH1, MSH2, ATM, ATR), p53 (CDKN2A, MDM2, MDM4, TP53), PI3K-AKT-mTOR signaling (PI3KCA, PIK3RA, PTEN, AKT1, AKT2, MTOR, RICTOR, TSC1, TSC2), Ras-Raf, MEK-Erk/JNK signaling (KRAS, HRAS, BRAF, RAF1, MAP2K1, MAP2K2, MAP2K4, MAPK1, MAPK3), and the RTK signaling family (EGFR, ERBB2, ERBB3, ERBB4, PDGFRA, PDGFRB, KIT, FGFR1, KDR). We analyzed the numbers of mutations with six functional and targetable pathways (Fig. 2D). The most frequently identified mutations were MYC in the cell cycle pathway, BRCA1 in the DNA damage response pathway, TP53 in the p53 pathway, PIK3CA in the PI3K-AKT-mTOR pathway, KRAS in the Ras-Raf-MEK-Erk/JNK pathway, and FGFR1 in the RTK signaling pathway.

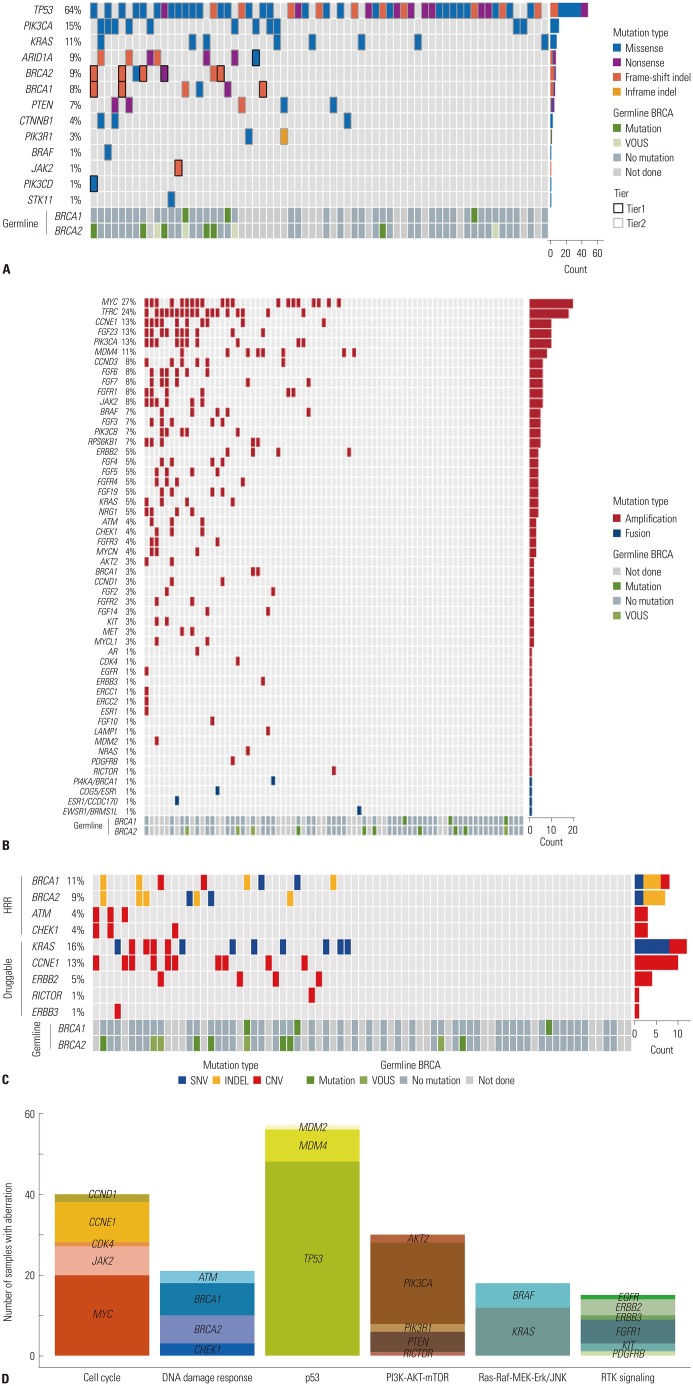
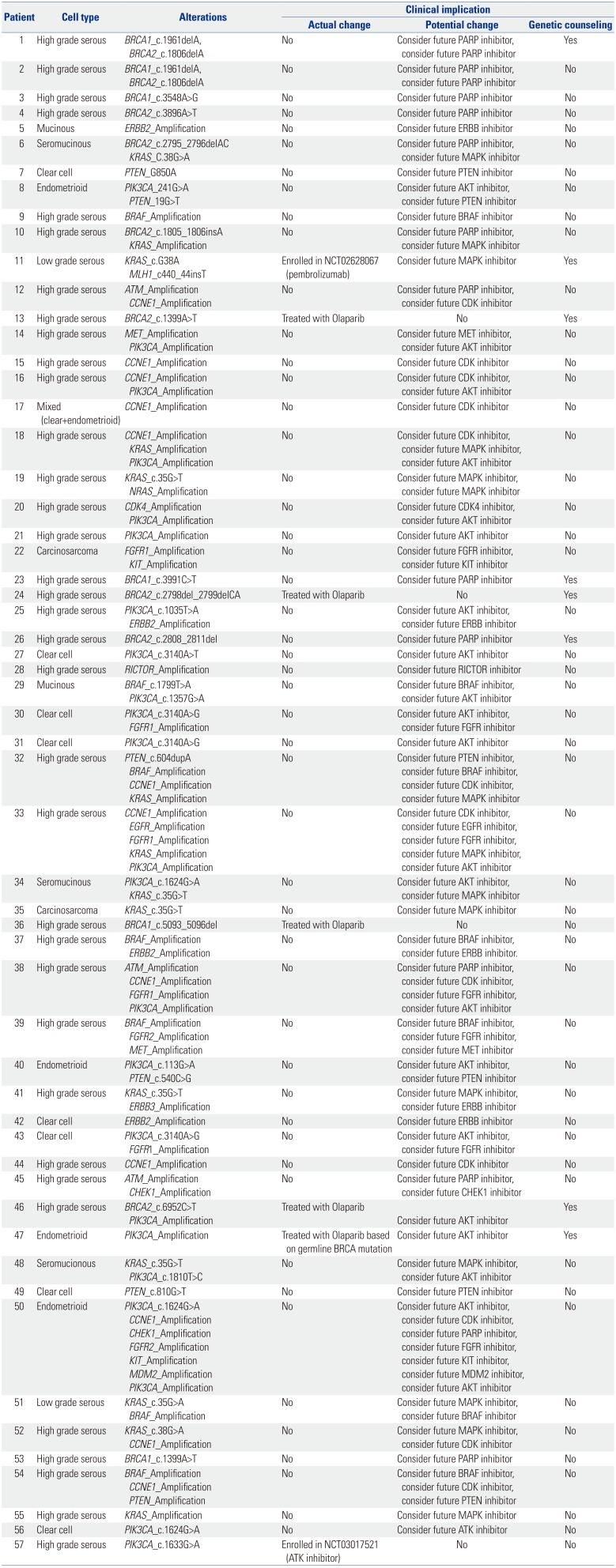
Fig. 1
Pie chart of the distribution of actionable somatic alterations. HRR, homologous recombination repair.
![]()
Fig. 2
Mutation, copy number variation profiling. (A) Single nucleotide variants and indels. Color legend of the variations represented, including frameshift indel, inframe indel missense, and nonsense. Vertical lines indicate gene names; horizontal lines indicate cases with germline mutations. (B) Copy number variations and fusions. Color legend of the variations represented, including amplification, fusion. Vertical lines indicate gene names; horizontal lines indicate cases with germline mutations. (C) Homologous recombination repair (HRR)-related genes and The Cancer Genome Atlas druggable genes. Color legend of the variations represented, including single nucleotide variants, indel, and copy number variations. Vertical lines indicate gene names; horizontal lines indicate cases with germline mutations. (D) Number of mutations with six functional and targetable pathways. Vertical lines indicate the number of cases with mutations; horizontal lines indicate specific genes grouped according to pathways.
![]()
Table 2
Tumor Molecular Profiles and Clinical Utility of Targetable Somatic Mutations
![]()
Targeted therapies
Of 57 patients with more than one actionable alteration, 7 (8.3%) were treated with matched therapies; 49 underwent standard chemotherapy without matched therapy; and 1 patient was treated with immunohistochemistry matched therapy (Fig. 3). Among the patients treated with matched therapies, five were treated with biomarker-driven therapy, and two were enrolled in biomarker matched clinical trials. BRCA1/2 mutations (n=5) were treated with poly ADP ribose polymerase (PARP) inhibitor; a MLH1 mutation with high microsatellite instability (n=1) was treated with a programmed cell death-1 (PD-1) inhibitor (ClinicalTrials.Gov Identifier: NCT02628067), and a PIK3CA mutation (n=1) was treated with an AKT inhibitor (ClinicalTrials.Gov Identifier: NCT03017521).

Fig. 3
CONSORT diagram. *ClinicalTrials. Gov Identifier: NCT03509246; †ClinicalTrials.Gov Identifier: NCT03414047. NGS, next generation sequencing; IHC, immunohistochemistry; PD-L1, programmed cell death-ligand 1.
![]()
Outcomes of matched therapy patients
A total of 7 patients were treated with a targeted agent, and the median value of prior lines of chemotherapy was 2 (range, 2–4). Of the 7 patients, five received PARP inhibitor therapy, one received PD-1 inhibitor, and one received AKT inhibitor therapy. Among patients who were treated with PARP inhibitor, four were treated with a PARP inhibitor for maintenance therapy, and one was treated with a PARP inhibitor as a 4th-line monotherapy. Among patients who underwent maintenance therapy with a PARP inhibitor, three were on follow-up without recurrence for more than 5 months; one experienced disease progression at 7 months after initiation of maintenance therapy. One patient treated with a PARP inhibitor as a 4th-line monotherapy had stable disease at the time of the analysis and had been undergoing treatment for 7 months. A patient treated with a PD-1 inhibitor experienced disease progression at 5 months after initiation of therapy. One patient treated with an AKT inhibitor had stable disease and had been undergoing treatment for 2 months.
Clinical impact
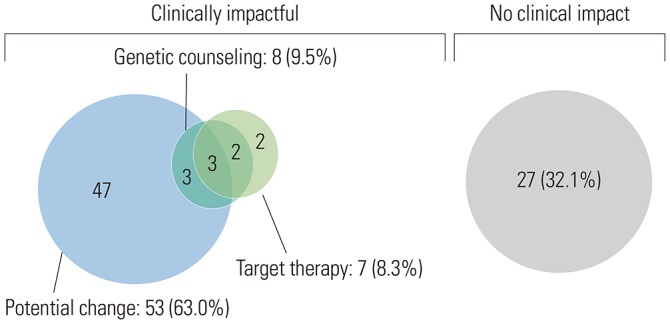
Clinically meaningful results are shown in Fig. 4. Clinically significant alterations were found in 57 (67.9%) patients. Of these 57, seven (8.3%) had matched targeted therapies, 53 (63.0%) had potentially actionable alterations, and eight (9.5%) and their atrisk family members without potentially actionable alterations received genetic counseling. Currently, the patients with potentially actionable alterations are currently either undergoing standard treatment or are in a state in which no disease is evident after treatment but remain candidates for matched therapy if there is a subsequent disease recurrence or progression. Several patients had multiple actionable alterations. These patients received matched therapy but had other alterations that may later make them potential candidates for matched therapy. Patients with actionable alterations that could make them future potential candidates for matched therapy are shown in Table 2.
DISCUSSION
In this study, we evaluated the clinical utility of NGS and identified clinically significant information beyond actionable alterations in ovarian cancer patients. In our review of NGS results for the 84 ovarian cancer patients in our institution, we found that 57 (67.9%) of them had one or more actionable alterations other than TP53 and that 16 (28.6%) of them had a mutation in HRR-related genes. Fifty-two (61.9%) patients had clinically significant alterations, seven (8.3%) were treated with matched targeted therapies, 48 (57.1%) had potentially actionable alterations, and eight (9.5%) received genetic counseling. In our study, 12 (14.3%) had germline BRCA1/2 mutations, and 50 (59.5%) had no germline BRCA1/2 mutation. In addition, the distribution of patients with somatic BRCA mutations at each time point of NGS were 8/47 (17.0%) in the chemo-naive group (pre-NAC, PDS) and 3/37 (8.1%) in the post chemotherapy group (IDS, relapse). Incorporation of NGS into standard clinical practice could provide a complementary tool with which to identify patients who might benefit from targeted therapies and genetic counseling.
NGS has been used in several studies to identify the actionable mutations of specific cancers and the clinical impact thereof.1112 Oberg, et al.11 showed the feasibility of incorporating NGS into pediatric hematology-oncology. They found it was clinically significant in 66% of all cases in which it was used. Its benefits included avoidance of inappropriate treatments, confirmation of definitive diagnoses, and identification of pharmacogenomics modifiers. Heong, et al.12 described the feasibility of a molecular screening program in Asian cancer patients. Eighty-two percent of all patients had at least one reportable genomic alteration. Eight percent of the patients with reportable alterations were treated with matched therapies based on their specific molecular alteration. Nine of these patients (45%; 95% CI, 23.1–68.5%) showed a clinical benefit, including three partial responses and six with stable disease. However, in the SHIVA trial,13 treatment with matched targeted agents based on a patient's actionable molecular alterations did not improve progression-free survival in heavily pretreated cancer patients, compared with their physicians' treatment choices. However, no studies have been conducted to assess the clinical impact, if any, of using NGS to identify actionable mutations in ovarian cancer patients.
In our study, 61.9% of all patients were identified as potential candidates for targeted therapy. This is comparable with other studies and may be the rationale for performing comprehensive genomic analysis in ovarian cancer patients. Despite our findings, only 8.3% of patients received targeted therapy. Even after their detection, matching susceptible mutations with specific efficacious treatment agents is especially difficult with ovarian cancer patients. Ovarian cancers have heterogeneous cell populations, an extremely complex etiology, and any number of mutations can occur as the cancer develops.14 As a further complication, health insurers are not obligated to cover the off-label use of expensive drugs.
Nevertheless, the incorporation of NGS into the treatment of ovarian cancer may have a significant clinical impact, including success in finding potential candidates for future targeted therapies and genetic counseling. In our study, 57.1% of all patients were potential candidates for future targeted therapy, and 9.5% of all patients received genetic counseling for the patient and their at-risk family members. In addition, the proportion of somatic BRCA mutations varied according to the time point of NGS analysis. Although the number of patients was small, the difference in the proportion of patients with somatic BRCA mutations in the two groups may be due to the effect of BRCA reversion by platinum-based chemotherapy. Reversion mutations in BRCA1/2 have been reported in ovarian cancer as a mechanism of acquired resistance to platinum-based chemotherapies and PARP inhibitors.1516 Based on these results, further studies are needed to analyze the genetic alterations in serial samples of chemo-naïve and post-chemotherapy patients.
Our study has some limitations. There are still cost-effective issues with NGS that can limit clinical testing and prevent or delay the initiation of targeted therapies, and failures, such as insufficient collection of tissues or improper sequencing, can lead to significantly increased turnaround times. In addition, our follow-up period on patients was too short to demonstrate definitively that matched targeted therapies based on the results of NGS yield better outcomes than empiric treatment choices.
NGS may help guide immediate and future treatment options for patients with ovarian cancer. Implementation of NGS served as a complementary tool to identify patients who may benefit from targeted therapies and genetic counseling. Further large-scale studies are needed to investigate the overall clinical utility and feasibility of NGS in ovarian cancer.
 XML Download
XML Download