

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
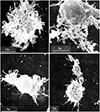
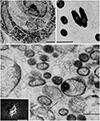
In 1993, I reported that Coxiella burnetii induces hairy cell (cbHC) transformation and that hairy cells (HCs) are observable in cases of polymorphic reticulosis infected with C. burnetii.1,2 To my knowledge, this was the first report suggesting that C. burnetii is associated with HC transformation. Morphologically, cbHCs were not differentiated from “HCs” described in hairy cell leukemia (HCL). The hairiness of original images of cbHCs and the intracellular presence of C. burnetii are shown in Figs. 1 and 2.1
C. burnetii is the agent of Q fever, or “query fever,” a zoonosis first described in 1937. C. burnetii has a cell wall similar to that of Gram-negative bacteria. This small coccobacillus (0.2 to 0.4 µm wide and 0.4 to 1 µm long) is an intracellular pathogen, replicating in eukaryotic cells. The estimated doubling growth time of the bacterium is between 20 and 45 h in cell culture.3 Recently, C. burnetii was found to be associated with B-cell non-Hodgkin lymphoma.4 In specific, reports of humans and animals HCL cases with Q-fever5,6,7 were valued. Those updated discoveries support my original proposition that C. burnetii induces HC transformation and warrant further discussion. Indeed, the significance of C. burnetii in cytoskeleton reorganization and apoptosis inhibition in cells infected with C. burnetii has garnered greater support.
HCL CHARACTERIZED BY CELLS WITH REORGANIZED CYTOSKELETONS
“Hairy cell” is a descriptive name proposed by Schrek and Donnelly in 1966.8 To date, HC has remained a diagnostic marker for HCL and variant HCL. The hairiness of cells has been also signified in variants of HCL lacking CD25, CD123, ANXA1, TRAP, and BRAF V600E expression.9 HC owes its name to the presence of numerous irregular projections protruding from the surface of cells, which are clearly visible in a phase contrast microscope and appear as irregular undulating ruffles or long villi when examined by a scanning electron microscope.10 HCL cells are able to cap surface immunoglobulins and concanavalin A receptors; the membrane redistribution of these structures is inhibited by cytochalasin B.11,12 Cytochalasin B inhibits both the rate of actin polymerization and the interaction of actin filaments in solution. HCL cells become firmly attached to culture dish surfaces and exhibit fibroblast-like projections and stellate features.13,14 Compared with normal B cells and other lymphomas, HCL specifically overexpresses β-actin (a non-muscle cytoskeletal isoform of actin).13,14
Cytoskeleton organization is aberrantly rearranged in the cells of B chronic lymphocytic leukemia, and F-actin was predominantly associated with dot-shaped structures scattered over the ventral membrane, representing spotty close contact adhesion sites analogous to “podosomes” described in other cell types.15 In HCL cells, polymerized actin (F-actin) is primarily found in the cortical cytoskeleton and supports the filamentous (F) membrane projections of HCL cells, whereas in normal B cells and in B-cell chronic lymphocytic leukemia cells, F-actin is primarily located in the central part of the cell.16 Thus, HCL cells apparently need to increase the expression levels of actin, concentrating it at the cell periphery, to sustain their prominent membrane projections.
HAIRINESS OF HCL IS IRRELEVANT TO BRAF-V600E MUTATION
In 2011, BRAF V600E mutation was observed in 100% of HCLs investigated.17 Since that observation was published, other groups18,19 have confirmed this result, establishing the paradigm that BRAF V600E is the disease-defining mutation of HCL. However, a few histologically, immunohistochemically, and clinically characteristic HCLs appear to lack the BRAF V600E mutation These includes the variant HCLs, splenic diffuse red pulp lymphoma and splenic marginal zone lymphoma.20,21,22 HCL cell lines expressing annexin A1 and displaying B-cell receptor signals characteristic of primary tumor cells lacking the signature BRAF mutation have also been reported: Although ANXA1 expression and other phenotypic markers together with cellular behavior showed some similarities with features typical of HCL in four cell lines (HCLL7876, Eskol, HC-1, and Hair-M), the absence of BRAF mutation placed into question their reliability as representative of this form of the malignancy.23 BRAF-V600E was also investigated in the human HCL cell lines BONNA-12, ESKOL, HAIR-M, and HC-1, but none of them carried BRAF-V600E mutation.24
BRAFV600E mutation occurring as early as in hematopoietic stem and progenitor B cells was reported in patients with HCL. In one study, BRAFV600E-mutant hematopoietic stem cells (HSCs) were transplanted into immunodeficient mice, and stable engraftment of BRAFV600E-mutant HSCs was verified. However, cells with classic HC–like morphology were not seen in the murine models.25 Since all animal procedures were conducted in accordance with the guidelines for the care and use of laboratory animals,25 the mice were immune deficient and free of pathogens, including C. burnetii. The hairless morphology of engrafted BRAFV600E-mutant HSCs in germfree mice may have been due to the absence of C. burnetii to induce cbHC. The same explanations are applicable for research with cultured cells in media containing various antibiotics. To evaluate any results observed in cell lines established ex vivo, an extra consideration is recommended, as significant differences in chromosome ploidy and in tumorigenicity between cells in glass (i.e., in vitro) and cells in plastic (i.e., in plastico) have been demonstrated.26,27 Also, cell lines established ex vivo may not represent their histologic origins.
Moreover, BRAF is a serine/threonine kinase that is commonly activated by somatic point mutations in human cancer, such as malignant melanoma and other solid tumors not signified by their hairy morphology. Presumptive BRAF mutations are found in normal tissue DNAs, cancer cell lines, colorectal cancers, gliomas, cancers, sarcomas, ovarian carcinomas, breast cancers and liver cancers.28 Thus, BRAF appears to be unrelated to the hairiness of cells, and stemness of BRAF mutants may not be specific to a certain disease.
C. burnetii AND ROUTE OF INFECTION
Most human infections occur after inhalation of infected aerosols of C. burnetii, and anecdotal cases of human-to-human transmission through infected aerosols have been reported after autopsies.29,30,31 Infection may occur after direct exposure to infected animals and their products (e.g., placenta, abortion products, hides, wool, manure, etc.), especially at the time of parturition or slaughtering.32,33
Ticks may play a role in the transmission of C. burnetii infection. This is illustrated by the detection of C. burnetii coinfection with other arthropod-borne pathogens in ticks.34 Q fever pneumonia is considered a nosocomial disease, and a recent case of respiratory nosocomial spread was reported. Because C. burnetii may persist for prolonged periods in the soil, these aerosols may also be produced long after the release of bacteria by infected animals. Moreover, bacterial aerosols can travel at least 30 km by the wind,35 resulting in Q fever cases far away from the primary source of contamination. Thus, Q fever cases are often diagnosed in persons with no recent contact with animals.
Birth products from infected parturient women are also a source of infection in obstetrical wards. A case of C. burnetii pneumonia was diagnosed by an obstetrician 7 days after he delivered an infant of an infected woman.36 Also, nosocomial transmission between two pregnant women sharing the same room has been reported.37 In these cases, the most probable source of the infection was infected, aerosolized vaginal excretion particles. C. burnetii infection through transfusion of blood collected from Q fever patients with bacteremia is also plausible, since the bacterium can survive in stored human blood samples.38
REORGANIZATION OF THE CYTOSKELETON BY C. burnetii
Many bacterial pathogens accomplish reorganization of the cytoskeleton by employing an arsenal of highly sophisticated mechanisms that subvert the cellular actin cytoskeleton to trigger their internalization into normally nonphagocytic host cells in order to escape the humoral immune defense.39 Among the multiple regulation steps of the actin cytoskeleton, bacterial factors interact preferentially with Rho GTPases.40
Reports of C. burnetii-induced morphologic changes in infected cells support the results of cbHC studies. Cytochalasin D, an inhibiter of actin polymerization, was found to prevent the uptake of C. burnetii without interfering with its binding. Thus, bacterial adherence may be necessary for tumor necrosis factor (TNF) production by monocytes. The monocyte αvβ3 integrin was shown to be involved in TNF synthesis, and interestingly, peptides containing arginine-glycine-aspartic acid sequences and blocking antibodies against αvβ3 integrin inhibited TNF transcripts induced by C. burnetii.41 Although both phase I and phase II variants engage αvβ3 integrin receptor in monocytes, dramatic reorganization of the F-actin cytoskeleton was observed with phase I bacteria. In human myelomonocytic cells infected with virulent C. burnetii, a profound polymorphic change in cells was reported.41
Differences in actin cytoskeleton reorganization according to the virulence of the infecting organism has been demonstrated, with virulent organisms showing lower reorganization than avirulent variants.42 In peripheral blood lymphocyte cultures infected with C. burnetii, exclusive pseudopodal extension and polarized distribution of F-actin were observed.43 Moreover, reorganization of the actin cytoskeleton by C. burnetii was reported to play a critical role in the internalization strategy of the organism, which is known to replicate exclusively within viable eukaryotic cells.44
A role for activation of protein tyrosine kinases (PTK) by C. burnetii has been implicated in actin cytoskeleton reorganization and bacterial phagocytosis. The phagocytosis of particles by macrophages was shown to depend primarily on reorganization of the actin cytoskeleton underlying the region of the plasma membrane that is in contact with particles.45 Virulent organisms were found to induce early PTK activation and tyrosine phosphorylation of several endogenous substrates, including Hck and Lyn, two Src-related kinases. Such PTK activation reflects C. burnetii virulence, since avirulent variants were unable to stimulate PTK.45 Actin cytoskeletal ruffling induced via Src tyrosine kinases was observed following the binding of phase I C. burnetii. This membrane ruffling required contact between C. burnetii and host cells and could be induced using purified lipopolysaccharide (LPS) from phase I bacteria.45,46
The ability to induce these ruffles appears to be dependent on the expression of Toll-like receptor 4 (TLR4) on the host cell surface.47 These observations suggest that LPS induces membrane ruffling and a type IV secretion system (T4SS) that temporally controls downstream signaling cascades after internalization. In animals, after aerosol transmission, C. burnetii targets alveolar macrophages and passively enters these cells by actin-dependent phagocytosis.48 The identification of a T4SS in C. burnetii indicated that this bacterium uses an active trigger mechanism (mediated by the secretion of T4SS effector proteins) to induce uptake after initial binding to the host cell.49 Although theT4SS of C. burnetii is not required for uptake by the host cell, it remains to be determined whether T4SS effectors contribute to the reorganization of the actin cytoskeleton during infection.50,51
Detailed molecular mechanisms are lacking due to difficulty in genetically manipulating these bacteria. Nevertheless, recent genetic breakthroughs have provided controllable systems by which bacteria-actin interactions can now be defined for this intriguing group of pathogens.52
In 2019, it was reported that C. burnetii affects E-cadherin (E-cad) expression. Researchers showed that C. burnetii severely impaired E-cad expression in circulating cells of Q fever patients. Cadherin switching is known to be a hallmark of neoplastic processes.53
In summation, these discoveries validate that C. burnetii reorganize the cytoskeletons of host cells, leading to the hairy appearance observed in cbHCs. Thus, cytoskeleton reorganization appears to be a C. burnetii-specific cytopathic effect that is not target specific.
Nature of HCL is B cell and peritoneal B1a cells were found to be permissive for virulent C. burnetii (Nine Mile phase I) strains in mice. C. burnetii infection B1a cells play an important role in regulating the C. burnetii infection-induced inflammatory response.54
INHIBITION OF APOPTOSIS BY C. burnetii
Eukaryotic cells often respond to intracellular pathogen invasion and stress induction by initiating the intrinsic apoptotic pathway as part of the innate immune defense.55 Two main pathways lead to apoptosis: the extrinsic cell death pathway is launched in response to stimulation of death receptor proteins at the cell surface by extracellular stimuli, while the intrinsic cell death pathway is initiated in response to intracellular stimuli.56 Apoptosis allows for pathogen clearance without inflammation and additionally leads to activation of the adaptive immune defense.57,58 As a countermeasure, intracellular pathogens have developed multiple mechanisms to inhibit host cell apoptosis.59
In 2009, researchers developed a means with which to axenically culture bacteria.60 These advances have provided the capacity to examine the pathogenesis of C. burnetii. More than a few studies have established that C. burnetii actively inhibits host cell death machinery by preventing the activation of both the intrinsic, mediated by mitochondria, and extrinsic, death receptor-mediated, apoptotic pathways. In particular, inhibition of cytochrome c release from mitochondria and continuous activation of the pro-survival kinases Akt and Erk1/2 have been observed during Coxiella infection.61,62,63
One of the first C. burnetii effectors to be identified, AnkG, was shown to inhibit apoptosis on a physiologically relevant scale.64,65 Further study revealed that AnkG interacts with the host mitochondrial protein p32 and that this interaction is important for transporting AnkG to the nucleus where it exerts an anti-apoptotic phenotype.65,66
Screening of previously identified C. burnetii effectors for the capacity to inhibit intrinsic apoptosis revealed two more anti-apoptotic effectors, CBU1524 and CBU1532, renamed CaeA and CaeB (C. burnetii anti-apoptotic effector).67 The discovery of three independent anti-apoptotic effectors indicates some functional redundancy; however, AnkG, CaeA and CaeB possess distinct mechanisms to block apoptosis and therefore may all contribute to the strong anti-apoptotic properties of Coxiella-infected cells. These studies, however, only screened a small proportion of known Coxiella effectors, and it is plausible that several more effectors contribute to maintaining host viability. Moreover, the data demonstrating the anti-apoptotic function of AnkG, CaeA and CaeB were not in the context of Coxiella infection. As such, it still remains to be determined whether the absence of these effectors, individually or in combination, impacts host cell survival during infection and the subsequent ability of Coxiella to replicate.
In 2019, Melenotte, et al.68 found the over-expression of genes involved in anti-apoptotic process and repression of pro-apoptotic genes as compared to samples from healthy donors by analyzing their gene expression by microarray.
COMPARABLE RISK FACTORS FOR COXIELLOSIS AND HCL
In HCL, an increased risk of the disease from farming exposure and a decreased risk from smoking exposure has been observed in studies independent of one another.69,70,71 Physicians practicing in rural areas should consider C. burnetii infection when patients present with atypical pneumonia and suggestive risk factors.72
SUMMARIZING REMARKS
C. burnetii reorganize the cytoskeleton and inhibits apoptosis of host cell. I propose these are C. burnetii induced cytopathic effects that are not target specific. Incidentally, a case of T-acute lymphoblastic leukemia with cells with unusual hairy projections was reported.73 Herein, C. burnetii infection of the patients should be considered.
 XML Download
XML Download