

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
A female patient, with the first known case of a SOX10 mutation variant causing Waardenburg syndrome type IV (WS4) without Hirschsprung's disease (HD), was born with meconium ileus. This led to multiple operations with complications that could have been avoided had the clinical picture of a Waardenburg syndrome (WS) variant been recognized earlier in the patient's hospital course. We describe the presentation, diagnostic workup, genetic findings, treatment, and complications of a patient with WS4 exhibiting chronic intestinal pseudo-obstruction (CIPO) instead of HD.
Go to : 
CASE REPORT
A premature female, born to unrelated Kuwaiti parents at a gestational age of 32 weeks, developed progressive abdominal distention with failure to pass meconium in the first two days of life. The patient had a white forelock, multidirectional nystagmus, white skin patches and repetitive extremity movements. A water-soluble contrast enema demonstrated a cut off at the sigmoid colon. The patient was found to have meconium ileus as well as malrotation during the exploratory laparotomy. A Ladd's procedure with saline irrigation and evacuation of meconium via an appendicostomy was performed. Of note, a suction rectal biopsy demonstrated the presence of ganglion cells as did the appendix. For two weeks postoperatively, the patient failed to pass stool and developed progressive abdominal distention and radiographic bowel dilation. Two contrast enemas showed an open colon but failure to reflux into the ileum. On postoperative day 18, the patient returned to the operating room for an enterostomy with closure after irrigation and evacuation of a recurrent meconium obstruction.
Over the next 8 weeks despite N-acetylcysteine via the nasogastric tube, the patient had intermittent bouts of abdominal distention and irregular stooling mostly with stimulation. She failed to reach full enteral feeds and required supplemental parenteral nutrition. A therapeutic water-soluble enema with N-acetylcysteine was performed nearly three weeks out from the second operation and another suction rectal biopsy was taken within 1 centimeter of the dentate line that again showed ganglion cells. Genetic testing for cystic fibrosis was negative. WS gene sequencing was in progress but the patient did fail an auditory brainstem response test.
Due to persistent meconium plugging, the patient underwent gastrostomy tube placement, ileostomy and mucous fistula creation and a full thickness rectal biopsy. The rectal biopsy again demonstrated ganglion cells, as did a portion of ileum. The ileal specimen did not have any glandular changes (viscous mucoid inspissation) typically seen with cystic fibrosis. Unfortunately, this operation was complicated by severe postoperative hypotension resulting in intestinal necrosis that required a massive intestinal resection with an open abdomen. Management options, including withdrawal of care, were discussed with the parents and they chose to proceed with further intervention. Once the patient stabilized, the duodenum just distal to the ampulla was stapled closed and resected secondary to necrosis. A segment of 10 cm of mid-jejunum was viable and left in discontinuity with both ends tied off and 8 cm of terminal ileum with the colon remained as a long Hartmann's. The abdomen was closed with an intra-abdominal drain placed at the duodenal stump.
At this point, the patient's gene sequencing revealed that she had WS4 with a SOX10 mutation. There was a heterozygosity found in the SOX10 gene for an undocumented sequence variant defined as c.895delC which resulted in a frameshift and premature protein termination (p.Gln299Serfs*12). This particular variant had not been previously reported. In addition, sweat testing was negative for cystic fibrosis.
Over the next four months, the patient underwent a tracheostomy for chronic respiratory failure and remained on total parenteral nutrition with a progressive decline in the amount of bilious drainage from the duodenal stump drain. Exploration with possible anastomosis and serial transverse enteroplasty (STEP) was then planned. Upon exploration, the proximal end of the isolated jejunal segment had fistulized to the end of the duodenum (Fig. 1A). The fistula was left in place, since an 18 French red rubber catheter was able fit through the fistula, and the distal aspect of the jejunum was anastomosed end-to-end to the terminal ileum. A STEP was performed which lengthened the mid-jejunal segment from 30 cm to 48 cm. This gave the patient 58 cm of small bowel from the head of the pancreas to the ileocecal valve (Fig. 1B). The entire colon was present and in continuity with no strictures.
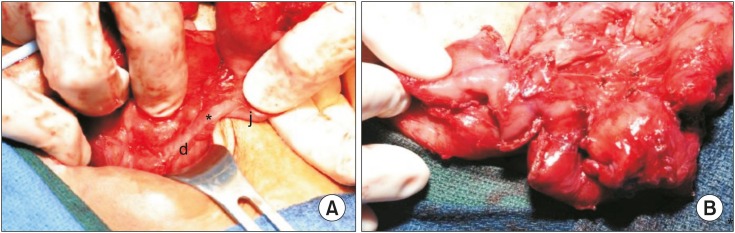
Postoperatively, the patient remained stable and developed no complications. Several weeks later, she was stooling with stimulation and able to resume feeds. WS4 workup included magnetic resonance imaging of the brain which demonstrated central hypomyelination. An electroencephalogram showed cortical slowing but no seizure activity and no correlation with her nystagmus or myoclonus. She was eventually discharged to a chronic care facility on supplemental parenteral nutrition tolerating low volume feeds and stooling a couple times per week with stimulation.
Go to :
DISCUSSION
WS is a neural crest disorder characterized by varying degrees of hearing loss and pigmentary changes of the hair (white forlock), eyes (heterochromia irides) and skin (white patches). WS results from the abnormal proliferation, survival, migration, or differentiation of pluripotent neural crest cells of the neural tube [12]. These cells develop into peripheral nervous system neurons, enteric neurons, craniofacial skeletal tissues, and melanocytes of the skin and inner ear [12]. Defects of these tissues during embryogenesis cause the clinically diverse features of WS which is subdivided into four major types [12].
Type I WS (WS1) refers to the first cases described by Waardenburg [3], a Dutch ophthalmologist and geneticist. These individuals have hearing loss and pigmentation abnormalities with widely spaced eyes and a broad nasal root [3]. Nearly all patients present with mutations of the transcription factor PAX3 (paired box gene 3) [2]. Type II WS is characterized by deafness and pigmentation defects without additional features and is caused by mutations in the MITF gene (microphthalmia-associated transcription factor) [2]. Type III WS, or Klein-WS demonstrates WS1 findings with extremity muscle hypoplasia and is also caused by PAX3 mutations [2].
Type IV WS, also referred to as Shah-WS or Waardenburg-HD (WS4) is characterized by WS1 features associated with the absence of distal enteric ganglia or HD [4]. Mutations of EDN3 (endothelin 3), EDNRB (endothelin receptor type B) and SOX10 are associated with WS4 [4]. SOX10, which belongs to the SOX (SRY-related high-mobility group [HMG] box) family of transcription factors, has been shown to play a role in the specification and differentiation of melanocytes, enteric progenitors and oligodendrocytes as well as glial cells of the peripheral nervous system [4]. Mutations occur in the heterozygous form and are most often de novo, with to date, just over 100 mutations identified in SOX10 [1]. These nonsense or frameshift as well as splice mutations in SOX10 remove all or part of the transactivation domain located in the C-terminal part of the protein and sometimes the HMG domain [5]. The patient presented in this case was found to have WS4 with a mutation in the SOX10 gene.
This patient's SOX10 mutation has not been previously reported in the literature. A heterozygosity was found in her SOX10 gene for an undocumented sequence variant defined as c.895delC. This is predicted to result in a de-novo frameshift causing a premature protein termination. These types of mutations in SOX10, which comprise about 50% of the WS4 etiology, are responsible for the WS4 variant syndromes PCWH (peripheral demyelinating neuropathy-central dysmyelinating leukodystrophy-WS-HD) or PCW (PCWH without HD) [15]. Interestingly, a few cases in the literature exhibit CIPO instead of HD [5], which correlates with our patient's clinical course as well as her genetic findings.
Several SOX10 mutations have been previously reported in patients displaying CIPO and WS features. Two of the reported patients had heterozygous de novo stop mutations (Q234X, amino acid change [700C>T, nucleotide change] and Q372X [1114C>T]) which removed the transactivation domains [4]. Both patients presented with intestinal obstructions as neonates with clinical courses similar to our case. Each had rectal and intestinal biopsies demonstrating normal ganglion cells and no histological abnormality. One of the patients presented with meconium ileus and required bowel washouts, stool softeners, frequent enemas and digital dilatation to maintain bowel function. The other patient passed away from severe neurological impairment with progressive hypoventilation and magnetic resonance imaging showing deficiency of myelination, which our patient has as well [4]. Pingault et al. [4] also identified a SOX10 mutation in a girl with CIPO, deafness and a peripheral demyelinating neuropathy without pigmentation abnormalities due to a de novo heterozygous frameshift mutation. A recent report of a SOX10 mutation in an Iranian WS4 family comprised of two affected siblings, their affected father and a healthy mother further demonstrates the variation in WS4 non-HD intestinal pathology [1]. The affected family members all had WS features with the son having HD and the father and 21-year-old daughter afflicted by chronic constipation. Anorectal manometry was used to rule out HD in the daughter. The father underwent no testing but gene sequencing of both siblings revealed a heterozygous variant c.422T>C of SOX10. Therefore, the same mutation might have demonstrated a different phenotype [1].
These cases, including our case, show that other intestinal pathologies, including CIPO, may be associated with WS and indicate that ganglion cell deficiency is not the only mechanism underlying the intestinal dysfunction of patients with SOX10 mutations [4]. Of note, to date, no CIPO has been described in association with EDNRB or EDN3 mutations [4] and no abnormality was found within these genes in our patient. CIPO is defined by repetitive episodes or continuous symptoms of bowel obstruction in the absence of a mechanical occluding lesion [46]. It differs from HD by the persistence of ganglion cells that appear normal, although a functional defect is possible and it may be neuropathic or myopathic in origin. Histopathological criteria are ill-defined [4] and CIPO may be related to intestinal neuronal dysplasia (IND) [7]. In addition, CIPO may present alone or in association with abnormalities of other organs. Urinary tract anomalies have been described in conjunction with CIPO as has intestinal malrotation [7]. Our patient did not have any urinary tract issues, but she did have malrotation. An interesting hypothesis for the association of malrotation and CIPO, by Devane et al. [8], postulates that impaired intestinal motor activity leads to inadequate rotation or vice versa that abnormal rotation interferes with intestinal nervous system formation. This hypothesis is possible considering our patient's presentation. The genetic basis for CIPO was uncertain until an autosomal dominant defect in the enteric smooth muscle actin g-2 (ACTG2) gene was identified in a Finnish family [910]. Unfortunately, our patient was not tested for this gene defect. Nevertheless, CIPO may present with symptoms and signs as well as a clinical and surgical course reminiscent of our case. Neonatal meconium ileus has been reported as an initial presentation of CIPO [6]. The reported patient was negative for cystic fibrosis and HD and received multiple laparotomies, an eventual gastrostomy and required enemas to stool, much like our patient [6]. In addition, CIPO patients may require ileostomy and small or large bowel resection [1011]. Our patient underwent an intestinal resection when she developed intestinal necrosis that was likely secondary to a low flow postoperative state related to the presence of a patent ductus arteriosis. This complication eventually led to the development of a duodenojejunal fistula. The occurrence of this type of postoperative spontaneous intestinal fistula has been reported in neonates with necrotizing enterocolitis [1213].
This case and the previously reported cases in the literature demonstrate that SOX10 gene sequencing is a consideration in WS patients without aganglionosis but with intestinal dysfunction [4]. In cases like our patient, CIPO may be a cause of intestinal pathology and therefore, with this knowledge, surgical management may be more accurately tailored in the setting of a SOX10 defect. For instance, earlier placement of a gastrostomy with an ileostomy may have negated the late complications of an entero-enteric fistula as well as short gut syndrome that developed in this patient. Further studies are needed to understand the defect underlying the pseudo-obstruction in SOX10 patients with WS features [4].
Go to :
 XML Download
XML Download