

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Progressive familial intrahepatic cholestasis (PFIC) is a group of severe genetic disorders that disrupt bile formation and present as cholestasis of hepatocellular origin, accounting for 10% to 15% of cholestatic liver disease in children [1]. PFICs are inherited in an autosomal recessive manner with estimated prevalence from 1/50,000 to 1/100,000 births [12]. PFIC is divided into three types: PFIC type 1, PFIC type 2, and PFIC type 3. PFIC type 1 is caused by a mutation in the ATPase phospholipid transporting 8B1 (ATP8B1) gene; this leads to impaired bile salt secretion, causing intrahepatic cholestasis, pruritis, failure to thrive, and progressive liver damage. PFIC type 2 is caused by a mutation in the gene that encodes the bile salt export pump (BSEP) protein, ABCB11, which plays a role in bile acid excretion. PFIC type 3 is caused by a mutation in the ABCB4 gene, that encodes multidrug resistance protein 3 and plays a role in phosphatidylcholine excretion; this mutation leads to impaired formation of mixed micelles that normally protect the biliary tree from detergent activity of bile salts [345].
In general, the defects in the mechanism of bile formation lead to end-stage liver failure in the first or second decade of life [2]. The prognosis varies among patients based on types of PFIC and variants of mutated genes; data on long-term outcomes are limited [4]. Due to the progressive nature of the disease, patients with PFIC often need liver transplantation [345]. In one study consisting of 13 patients with PFIC type 1 and 39 with PFIC type 2, one-third of the patients underwent liver transplantation [6].
PFIC type 1 (OMIM #211600) is associated with ATP8B1 gene mutations located on chromosome 18q21-q22, that encodes the FIC1 protein [7]. The FIC1 protein is a P-type ATPase thought to act like an aminophospholipid transporter that induces movement of phosphatidylserine and phosphatidylethanolamine from the outer leaflet to the inner leaflet of the plasma membrane of hepatocytes. This protein is postulated to play a role in maintaining asymmetric distribution of lipids in the phospholipid bilayer, which protects the canalicular lumen against high bile salt concentrations [4].
Despite substantial prevalence of PFIC type 1 among Caucasian children with neonatal cholestasis, PFIC type 1 is rare in East Asia. To our knowledge, fewer than 20 patients with PFIC type 1 have been identified in East Asia [8910]. We report a case of novel compound heterozygous mutations (p.Glu585Ter and p.Leu749Pro) in the ATP8B1 gene in an infant presented with liver failure.
CASE REPORT
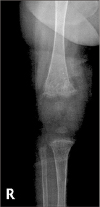
A seven-month-old female presented with a six-month history of cholestasis was transferred to a tertiary hospital for evaluation of intrahepatic cholestasis and recurrent diarrhea. She was born at gestational age 40 weeks, weighing 3.2 kg via cesarean section. Patient's weight and height were 6.18 kg (3rd percentile) and 65 cm (<3rd percentile), respectively. Physical examination revealed three-finger-breadth hepatomegaly without splenomegaly or ascites. Radiographic examinations revealed rickets with secondary fractures in bilateral distal femurs (Fig. 1). Laboratory testing revealed normal complete blood count, urea, creatinine, and electrolytes. Serum aspartate aminotransferase was 331 IU/L; alanine aminotransferase, 747 IU/L; total bilirubin (TB), 25.0 mg/dL; and direct bilirubin, 22.6 mg/dL. The γ-glutamyl transferase (GGT) level was 23 IU/L (normal range, <36 IU/L). Prothrombin time was 10.6 seconds (international normalized ratio, 0.93) and partial thromboplastin time was 29.8 seconds. Serum albumin was 2.9 g/dL and cholesterol levels were within reference range. Despite frequent fatty stool, fecal fat screenings by Sudan III staining were negative. Fecal elastase was 135.4 μg/g (normal range, >200 IU/L). Liver ultrasound revealed mild hepatomegaly with no evidence of bile obstruction. Liver biopsy at age seven months revealed moderate cholestasis and mild portal inflammation with focal bile duct absence and periportal and perisinusoidal fibrosis.
Gene study revealed novel compound heterozygous mutations of c.1753G>T, p.E585* and c.2246T>C, p.L749P in the ATP8B1 gene (Fig. 2). Parents are nonconsanguineous and healthy with heterozygous variants in ATP8B1. Variants were detected in neither the Exome Aggregation Consortium (ExAC) nor 1000 Genomes. Based on the guidelines of the American College of Medical Genetics and Genomics (ACMG) [11], E585* variants were pathogenic. The other p.L749P variant is of unknown significance, at highly conserved loci (PhastCons, 0.99) and reveals deleterious predictions (SIFT, 0.01 and PolyPhen-2, 0.993).
 | Fig. 2Variants of the ATP8B1 gene in the patient. The patient inherited a missense mutation from her mother (L749P) and a nonsense mutation from her father (E585X).
|
Patient underwent cadaveric donor liver transplantation at age 19 months. Biopsy of the extracted liver also revealed mild portal inflammation with periportal fibrosis and loss of bile ducts with ductular reaction, consistent with PFIC type 1 (Fig. 3). BSEP expression was intact and CD10 expression was not identified along the bile canaliculi. On day 82 posttransplant, the patient presented elevated aspartate and alanine aminotransferase levels, ascites, and PT prolongation. Liver biopsy revealed severe fatty change with mild portal inflammation and severe bile duct damage and bile ductular proliferation (Fig. 4).
 | Fig. 3Biopsy of the explanted liver. (A) Mild portal inflammation with focal bile duct absence, periportal and perisinusoidal fibrosis, and moderate cholestasis (H&E, ×400). (B) Bile salt export pump (BSEP) protein was normally distributed on immunohistochemistry (IHC) staining of the patient's liver (IHC with anti-BSEP) (H&E, ×400). (C) Decreased CD10 staining suggested fewer expressed canaliculi (IHC with CD10) (H&E, ×400). (D) Dilated canaliculus with coarse and granular bile and intraductal bile plugs (electron microscopy) (H&E, ×4,000).
|

DISCUSSION
This is the first report of PFIC type 1 with novel compound heterozygous mutations in Korea. Consistent with previous observations of clinical features of patients with PFIC type 1, our patient had hyperbilirubinemia due to intrahepatic cholestasis, normal serum GGT values, and pancreatic insufficiency. In addition, liver biopsies were consistent with previously reported pathological findings such as mild portal inflammation with periportal fibrosis, loss of bile ducts, and intact BSEP expression. In addition, the patient developed severe post-transplant steatohepatitis, often reported in patients with PFIC type 1 [12].
PFIC type 1 is generally diagnosed by clinical and laboratory parameters. PFIC type 1 patients typically present with recurrent intrahepatic cholestasis, pruritis, and failure to thrive. Other manifestations include pancreatic insufficiency, sensorineural deafness, hepatosplenomegaly, and lipid-soluble vitamin deficiency [3]. Laboratory findings generally reveal conjugated hyperbilirubinemia with normal serum GGT levels. However, diagnostic criteria for PFIC type 1 are not well established. Less severe phenotype of PFIC type 1 is benign recurrent intrahepatic cholestasis (BRIC), of which diagnostic criteria was proposed by Luketic and Shiffman [13]: (1) two of more episodes of jaundice separated by a symptom-free interval lasting several months to years (at least six months), (2) laboratory values indicating intrahepatic cholestasis, (3) severe pruritis secondary to cholestasis, (4) liver histology consistent with centrilobular cholestasis, (5) normal intra and extrahepatic bile ducts confirmed by cholangiography, and (6) absence of factors associated with cholestasis. PFIC type 1 differs from BRIC in that the disease eventually progresses to biliary cirrhosis. Liver biopsy of a PFIC type 1 patient reveals bland cholestasis with coarse and granular bile [5] and can provide diagnostic clues using electron microscopy and immunohistochemistry. However, currently, there is no confirmatory test for PFIC commercially available for clinical practice.
We identified novel compound heterozygous mutations in the ATP8B1 gene (c.1753G>T, p.E585* and c.2246T>C, p.L749P). Non-sense mutation p.E585* variant is on exon 15 and similar mutations on exon 15 (p.R525*) and 18 (p.E665*) were reported in other patients with PFIC type 1 [614]. The p.L749P variant is deleterious in in-silico predictions and is in a highly conserved location. The p.L749P mutation is likely be a pathogenic variant, with regard to typical phenotypes of PFIC type 1 and post-transplant steatohepatitis, according to ACMG guidelines. Verification of pathogenicity of candidate variants of the ATP8B1 gene is difficult because functional studies are complex. In addition, exact mechanisms of cholestasis in PFIC have not been fully elucidated. It is postulated that altered protein function may disturb bile acid secretion. In addition, down-regulation of the farnesoid X receptor leads to down-regulation of BSEP protein, up-regulation of bile acid synthesis, and up-regulation of intestinal apical sodium bile salt transporter, leading to increased bile salt concentration in hepatocytes [115]. Increase in glycine- and taurine-conjugated hydrophobic bile salts over the threshold may damage cell organelles such as mitochondria and consequently, cause hepatocellular damage [16].
Genetic profiles are well known in the ATP8B1 gene (Table 1 [68141718192021]). Generally, nonsense, frame-shift, and large deletion mutations have been seen commonly in PFIC type 1 patients (13%, 26%, and 3%, respectively) in contrast to missense mutations (58%) seen in BRIC [14]. However, only a few mutations were known among children in East Asia. Chen et al. [8] reported four Taiwanese patients with infantile-onset chronic intrahepatic cholestasis with ATP8B1 mutation. Hori et al. [9] reported a center experience of living donor liver transplant in PFIC type 1 patients spanning two decades, comprising 11 patients. Numakura et al. [10] reported a PFIC type 1 patient with novel compound heterozygous mutation of ATP8B1, expected to receive a partial external biliary diversion.
Table 1
ATP8B1 mutations

| Variant types | Nucleotide change | Amino acid change | Inheritance | Reference |
|---|---|---|---|---|
| Deletion | g.24774-42062del | Truncated protein | Homozygote | [6] |
| Missense | c.625 C>A | p.P209T | Homozygote | [6] |
| Frameshift | c.589_592inv;592_593insA | NA | Homozygote | [17] |
| Splicing | c.625+5 g>t | NA | Homozygote | [6] |
| Splicing | c.782-1G>A | NA | Compound heterozygote | [6] |
| Missense | c.923 G>T | p.G308V | Homozygote | [18] |
| Missense | c.1286 A>C | p.E429A | Heterozygote | [14] |
| Missense | c.1336 G>A | p.G446R | Compound heterozygote | [8] |
| Nonsense | c.1573 C>T | p.R525X | Compound heterozygote | [6] |
| Deletion | c.1587-1589 del CTT | p.del F529 | Compound heterozygote | [14] |
| Missense | c.1798 C>T | p.R600W | Homozygote | [14] |
| Missense | c.1982 T>C | p.I661T | Compound heterozygote | [18] |
| Nonsense | c.1993 G>T | p.E665X | Compound heterozygote | [14] |
| Skipping | c.2097+2 T>C | Deletion of exon 18 | Homozygote | [8] |
| Deletion | c.2097+2 T>C | p.del I645-I699 | Compound heterozygote | [18] |
| Missense | c.2103 G>C | p.G702R | Compound heterozygote | [6] |
| Missense | c.2150 C>A | p.T717N | Homozygote | [6] |
| Nonsense | c.2271 T>A | p.Y757X | Compound heterozygote | [8] |
| Missense | c.2855 G>A | p.R952Q | Homozygote | [14] |
| Missense | c.2854 C>T | p.R952X | Heterozygote | [14] |
| Missense | c.2982 C>A | p.S994R | Compound heterozygote | [6] |
| Missense | c.3035G>T | p.S1012I | Heterozygote | [19] |
| Frameshift | c.3623dupGCTCGGC | p.T1209Lfs28X | Compound heterozygote | [6] |
| Nonsense | c.1993 G>T | p.E665X | Homozygote | [14] |
| Missense | c.1660 G>A | p.D554N | Homozygote | [14] |
| Missense | c. 3118 G>A | p.G1040R | Homozygote | [14] |
| Splicing | IVS8+1G>T | NA | Homozygote | [14] |
| Frameshift | 3622_3628delGCCTACG | p.A1208fs | Homozygote | [14] |
| Missense | c.2674G>A | p.G892R | Homozygous | [18] |
| Missense | c.863T>C | p.L288S | Homozygous | [18] |
| Deletion | c.NT2384 | p.795delGNR | Homozygous | [18] |
| Skipping | c.IVS23-3C>A | Deletion of exon 24 | Homozygous | [14] |
| Missense | c.2599C>T | p.867C | Heterozygous | [20] |
| Missense | c.1367C>T | p.T456M | Compound heterozygote | [21] |
| Nonsense | c.1804C>T | p.R602X | Compound heterozygote | [21] |
| Missense | c.380T>C | p.L127P | Homozygote | [14] |
| Missense | c.1235 G>C | p.R412P | Homozygote | [14] |
| Splicing | c.IVS3+1_+3delGTG | NA | Homozygote | [14] |
| Splicing | c.279 G>A | p.A93A | Homozygote | [14] |
| Frameshift | c.2124_2125insGAGCTACAGCTATTGAAGGC | K709fs | Homozygote | [14] |
| Splicing | c.IVS21+5G>A | NA | Homozygote | [14] |
| Frameshift | c.2543_2556dupAGCGGCAGAAAAAC | p.F853fs | Homozygote | [14] |
| Splicing | c.IVS17-1 G>A | NA | Homozygote | [14] |
| Splicing | c.IVS18+2 T>C | NA | Homozygote | [14] |
| Missense | c.1604A>T | p.H535L | Homozygote | [14] |
| Frameshift | c.2596_2599dupTGCC | p.R867fs | Homozygote | [14] |
| Nonsense | c.3040C>T | p.R1014X | Homozygote | [14] |
| Missense | c.2063A>G | p.D688G | Homozygote | [14] |
| Splicing | c.IVS3-2 A>G | NA | Compound heterozygote | [14] |
| Frameshift | c.2016delG | p.K672fs | Compound heterozygote | [14] |
| Frameshift | c.2873delA | p.N958fs | Compound heterozygote | [14] |
| Nonsense | c.2788 C>T | p.R930X | Compound heterozygote | [14] |
| Frameshift | c.3069_3070delAA | p.Q1023fs | Compound heterozygote | [14] |
| Missense | c.1208 C>A | p.S403Y | Compound heterozygote | [14] |
| Frameshift | c.2373_2374insT | p.792fs | Compound heterozygote | [14] |
| Frameshift | c.614_615insA | p.N205fs | Compound heterozygote | [14] |
| Missense | c.2197 G>A | p.G733R | Heterozygote | [14] |
| Missense | c.1498 T>C | p.Y500H | Heterozygote | [14] |
| Missense | c.1371 G>A | p.G457G | Heterozygote | [14] |
![]()
Current medical treatment for type 1 PFIC is limited, as most of the medications used, including ursodeoxycholic acid [22], rifampin, and cholestyramine, provide only symptomatic relief and have uncertain effects on disease progression. Some patients may experience symptom relief or even slower disease progression after biliary diversion [1]. Growth failure is observed in 95% of these patients, due to cholestasis, hepatic failure, and fat malabsorption [23]. Most of these patients also have rickets and osteopenia from impairment of vitamin D absorption and calcium metabolism [23]. Liver transplantation is associated with development or aggravation of diarrhea because larger amounts of bile acid reach the intestine [1224]. Also, Lykavieris et al. [12] proposed that liver transplantation does not improve catch-up in height velocity or quality of life, and may cause appearance of liver steatosis or in some cases progression to cirrhosis [25]. The patient in this report also remains under the third percentile range in weight and height and has developed liver steatosis.
In conclusion, we report the first Korean case of PFIC type 1, which is highly unusual clinically and genetically. Pathogenicity of these novel compound heterozygous mutations of p.E585* and p.L749P will be confirmed by additional identification of patients with the same variants.
 XML Download
XML Download