

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
We report 9-year-old fraternal twins from a family with piebaldism, having congenital depigmented macules and meeting the diagnostic criteria for neurofibromatosis type 1 (NF1) based on the presence of multiple café-au-lait macules (CALMs) and intertriginous freckling at the same time. In the literature, there are eight case reports describing twelve patients with similar cutaneous findings. Most of them have been attributed as an overlap of piebaldism and NF112345678. No systemic findings of NF1 were observed in any of these reports except one patient2. And no NF1 gene mutation was detected24678. Therefore, it's still a debatable issue that CALMs and intertriginous freckling may be seen in the clinical spectrum of piebaldism or these patients should be regarded as coexistence of piebaldism and NF1 and monitored closely for NF19101112.
CASE REPORT
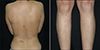
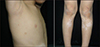
We present 9-year-old fraternal twins (a boy and a girl) with piebaldism, CALMs and intertriginous freckling. The mother of them reported that both of the patients had depigmented macules including hyperpigmented skin areas that were present at birth and showed no changes over time. It was also stated that small brown spots became apparent in the axillary and inguinal folds in infancy and they increased in number and size and spread over the body during childhood. Their physical and mental developments were normal. Family history revealed inheritance of piebaldism on the maternal side. Their mother had also similar findings (Fig. 1), and she mentioned that her sister and aunt had depigmented macules, frontal white forelock and multiple CALMs (Fig. 2). However, there were no neurofibromas or other clinical findings of NF1 in any members of family.
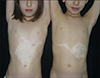
The boy had irregular bordered depigmented macules in different sizes including hyperpigmented skin areas on his ventral trunk, left leg and both arms. He had also numerous CALMs, at least 15 of which were >5 mm in diameter, and bilateral axillary and inguinal freckling (Fig. 3). Similarly, the girl had depigmented macules including islands of hyperpigmented macules on her knees, in the midline of the ventral trunk and on right arm. She had also numerous CALMs, 9 of which were >5 mm in diameter, and bilateral axillary and inguinal freckling (Fig. 4). We received the patient's consent form about publishing all photographic materials.
No other findings such as poliosis or neurofibromas were detected in neither of the twins. There were no pathologic findings on ophthalmic, neurological, and audiological examinations. Routine laboratory tests, skeletal survey, chest radiography, and magnetic resonance imaging of the temporal bone and brain were normal. Minimal mitral insufficiency was detected in boy and minimal mitral valve prolapse was determined in girl by echocardiography. Genetic testing for NF1 didn't demonstrate NF1 gene mutation in neither of them. Because of the financial reasons genetic testing for KIT and SPRED1 mutations couldn't be performed.
DISCUSSION
Piebaldism is a rare autosomal dominant genodermatosis characterized by congenital depigmentation in affected areas of the skin and/or hair113. Genetic studies identified that clinical manifestations and phenotypic severity of piebaldism correlate with the type and localisation of the KIT gene mutation13. The prevelance of NF1, another autosomal dominant genodermatosis, is higher than piebaldism. The different clinical features of NF1 are CALMs, intertriginous freckling, neurofibromas, Lisch nodules, and increased malignant and benign tumors. Based on the National Institutes of Health Diagnostic Criteria. The presence of six or more CALMs over than 5 mm in diameter in prepubertal individuals and intertriginous freckling as in our patients seems to be enough for the diagnosis of NF1. CALMs and intertriginous freckling are apparent at birth and childhood while the other diagnostic signs are uncommon in small children. Because of the fact that the reliability of the diagnostic criteria improves in advancing ages, the diagnosis of NF1 especially in small children is difficult to be made in certainty. Therefore, prepubertal patients with CALMs and intertriginous freckling should be closely followed in terms of other findings of NF11415. Because none of the relatives on the maternal side with piebaldism had other signs of NF1, the presence of CALMs and the intertriginous freckling in our patients may be a rarely seen clinical variant of piebaldism rather than the diagnostic criteria of NF1.
Piebaldism is characterized by the depigmented macules of the skin owing to the absence of melanocytes while, NF1 is characterized by hyperpigmented macules, intertriginous freckling and pigmented iris hamartomas depending on the increased number of melanocytes and macromelanosomes. Piebaldism is associated with the mutations of the KIT (receptor tyrozine kinase) gene on chromosome 4q12 or the SLUG (zinc finger neural crest transcriptional factor) gene on chromosome 8q11, whereas NF1 is associated with mutations of the NF1 gene on chromosome 17q11.2. Inactivating mutations or deletions of the KIT or SLUG genes result in decreased receptor tyrosine kinase signaling and a decrease in melanogenesis, but the mutations or the deletions of the NF1 results in enhanced receptor tyrozine kinase signaling. In some studies, increased KIT expression and activity have also been demonstrated on NF1-associated neurofibromas69.
The absence of other clinical findings of NF1 in patients with similar phenotypes and the absence of NF1 mutations in a small number of patients undergone genetic testing suggest that these findings are a rare phenotype of piebaldism. Recently, Spritz10 claimed that multiple CALMs and intertriginous freckling occur frequently in the typical patients of piebaldism. Spritz10 also reported that these hyperpigmented macules are the minor clinical diagnostic criteria for NF1. For this reason it has been suggested that in such patients the diagnosis of NF1 must be based on the nonpigmentary diagnostic criteria (e.g., neurofibromas) and demonstration of a mutation in NF1 gene by DNA testing10. Subsequently, Duarte et al.11 suggested that there is no definition reported by Spritz as “minor clinical diagnostic criteria for NF1” and that distinction is only a personal opinion. It has been also claimed that occurrence of neurofibromas, which are mentioned as necessary for the diagnosis of NF1, is not possible in these patients with piebaldism due to the KIT gene mutation. Although the sensitivity of the molecular recognition of NF1 is 95%, it has also been reported to be impossible to detect microdeletions which accounts for 5%~10% of NF1 mutations. Therefore, it has been thought that NF1 diagnosis cannot be excluded in these patients and a possible NF1 diagnosis should not be missed in order to prevent systemic complications in later periods11.
Although comprehensive genetic analyses were not performed in the prior reported patients, KIT gene mutation has been detected in subsequent five patients while there was no NF1 gene mutation in any of them678. Recently, the loss of function of the SPRED1 (Sprouty-related, Ena/vasodilator-stimulated phosphoprotein homology-1 domain containing protein 1) gene associated Legius syndrome with NF1-like clinical findings has also been investigated in three patients with piebaldism, multiple CALMs and intertriginous freckling78. Chiu et al.7 identified a noval heterozygous mutation in the KIT gene, resulting in an aminoacid substitution in the intracellular tyrosine kinaz domain, in a 5-year-old girl. It has been thought that this different mutation in the KIT gene leads to inadequate phosphorylation of SPRED1 gene on chromosome 15q, resulting loss of inhibition of the ras/MAPK pathway, with multiple CALMs and clinical phenotype similar to Legius syndrome7. The other features of Legius syndrome include lipomas, macrocephaly and neurocognitive disorders. Although the patients with Legius sydrome meet the diagnostic criteria for NF1, they do not need the careful clinical follow-up for NF1 because of no neurofibromas, Lisch nodules and central nervous system tumors are seen78. In the other two patients reported by Stevens et al.8, complex KIT gene mutations were defined whereas no mutation was detected in the NF1 and SPRED1 genes.
Consequently, it has been determined that all patients regarded as coexistence of piebaldism and NF1 had no other findings of NF1 except one patient and there was no mutation of NF1 gene in genetically analyzed patients. In addition to this, as in our patients, it is not possible to exclude a possible diagnosis of Legius syndrome in these patients, because the SPRED1 gene mutation analysis was not performed in the majority of prior reported ones. Chiu et al.7 claimed that the coexistence of piebaldism and Legius sydrome would be much less than the association of NF1 because both KIT and SPRED1 gene mutations are very rare and each is on different chromosomes. However, the possibility of these recently reported mutations in our patients cannot be excluded due to the lack of comprehensive genetic testing.
As a summary, we identified two patients from a family with piebaldism, associated with multiple CALMs and intertriginous freckling. We consider that this clinical presentation is an uncommon phenotypic variant of piebaldism due to absence of NF1 mutation and the other features of NF1 disease or Legius syndrome. For this reason, we think that these patients do not need a careful clinical follow-up and further examination for NF1. Besides, it may be suitable that these individuals with piebaldism showing a clinical phenotype similar to NF1 or Legius syndrome should be further tested for KIT and SPRED1 gene mutations.

 XML Download
XML Download