

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Most transient receptor potential (TRP) channels are activated by trimeric G protein signaling, which increases cytoplasmic Ca2+ concentrations [12] and generates lipid second messengers following hydrolysis of phosphatidylinositol 4,5-biphosphate (PI(4,5) P2) [345]. Also, the channels are directly activated by Gα [678] and Gβγ subunits [9]. Of the TRP superfamily, canonical TRP proteins, especially transient receptor potential canonical (TRPC) 4 and TRPC5 form Ca2+-permeable cation channels activated after stimulation of the phospholipase C (PLC) pathway [10]. Other factors have also been shown to affect the activity of TRPC channels. These include the activated Gαi2 [68] and Gαq [11], Ca2+ and PI(4,5)P2.
For TRPC subunits, TRPC4 and TRPC5 form homomeric channels, whereas TRPC1 forms heteromeric channels with TRPC4 or TRPC5 [1213]. By forming heteromers, TRPC1 changes the biophysical properties of homomeric channels from a double-rectifying shape to an outward-rectifying current-voltage shape. Because TRPC1 is broadly distributed in native tissues, studies for heteromeric TRPC1/4 and TRPC1/5 channels are more physiologically important than homomeric TRPC4 and TRPC5. The TRPC1 subunit only translocate to the plasma membrane when TRPC4 or TRPC5 is coexpressed. Co-immunoprecipitation data proposed that combination of TRPC1–TRPC4, TRPC1–TRPC5 form a heteromeric complex [11]. Furthermore, interaction regions between the subunits were identified using the fluorescence resonance energy transfer technique [14].
The Gαq subunit activates phospholipase C β (PLCβ) and in turn hydrolyzes PI(4,5)P2 to diacylglycerol (DAG) and inositol trisphosphate (IP3). The downstream from the PI(4,5)P2 breakdown, DAG activates protein kinase C (PKC), while IP3 triggers Ca2+ release from stored endoplasmic reticulum (ER) Ca2+. Previously, we suggested a novel mechanism of self-limiting activation of TRPC1/4 and TRPC1/5 channels by Gαq protein coupled receptor stimulation [11]. Our previous work showed that the heteromeric channels directly interact with activated Gαq, and subsequent PI(4,5)P2 hydrolysis induces inactivation of the channels. In signal transduction cascade downstream of PLCβ, the dependence of TRPC1/4 and TRPC1/5 function on Ca2+ or PKC activation is not well known. In contrast, roles of IP3R activation, PKC and Ca2+ on TRPC4 and TRPC5 channels are relatively distinct [15161718].
The study aimed to determine the downstream mechanism behind PLCβ-induced regulation, exploring the effects of Ca2+ and PKC activation on the gating of TRPC4 and TRPC5 channels. In the present study, we showed that the inactivation process of TRPC1/4 and TRPC1/5 currents can be facilitated by Ca2+ release from the ER and activation of PKC, respectively. These findings provide a molecular mechanism for gating of heterotrimeric TRPC1/4 and TRPC1/5 channels to allow control of Ca2+ influx to prevent Ca2+-dependent cell toxicity.
METHODS
Cell culture and transfection
Human embryonic kidney (HEK293) cells (ATCC, Manassas, VA, USA) were maintained according to the supplier's recommendations. HEK293 cells were incubated in Dulbecco's Modified Eagle's Medium supplemented with 10% heat-inactivated fetal bovine serum, penicillin (100 U/ml), and streptomycin (100 µg/ml) at 37℃ in a 5% CO2 humidified incubator. Cells were seeded in confocal dish for recording images or a 12-well plate for whole-cell patch clamp recordings, and 6-well plate for western blot. The following day, transfection was carried out by using the FuGENE 6 Transfection Reagent (Promega, Madison, WI, USA) and Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. All experiments were performed 20–30 h after transfection.
Solutions and drugs
The patch pipettes were made from borosilicate glass and had resistances of 3–5 MΩ when filled with standard intracellular solution; 140 CsCl, 10 N-2-hydroxyethylpiperazine-N′-2-ethanesulphonic acid (HEPES), 0.2 Tris-GTP, 0.5 ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA), 3 Mg-ATP, pH 7.3 with CsOH (in mM). Additional 1,2-bis-(2-aminophenoxy) ethane-N,N,N′,N′-tetraacetic acid (BAPTA) concentrations in the pipette solutions are stated in figure legend. External solution was perfused constantly as follow; 135 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 glucose,10 HEPES, pH 7.4 with NaOH (in mM).
Electrophysiology
The cells were transferred onto a solution chamber on the stage of an inverted microscope (IX70; Olympus, Tokyo, Japan). Whole cell configuration was used to measure TRPC channel currents in HEK293 cells as described previously [619]. Cells were attached to coverslip in the small chamber for 10 min prior for the patch recording. The currents were recorded using an Axopatch 200B patch-clamp amplifier (Axon Instruments, Foster City, CA, USA). Voltage ramp pulse from +100 mV to −100 mV over period of 500 ms was imposed with a −60 mV holding membrane potential. Experiments were performed at room temperature (18℃–22℃). The recording chamber was continuously perfused at a flow rate of 1–2 ml/min.
Microscopic image acquisition
HEK293 cells were cultured in a 35-mm coverslip bottom dish or a 12-well plate to obtain. To obtain the image of a cell, we used an inverted microscope with 60× oil objective lens. Images were acquired on the cooled 3 MHz (14 bit) EMCCD camera (iXon Ultra 888; ANDOR, Belfast, UK) with 1 × 1, 2 × 2, or 3 × 3 binning under the control of MetaMorph 7.6 software (Molecular Devices, Sunnyvale, CA, USA). Each images were exposed over period of 100 ms.
Calcium imaging
The ratiometric measurement of [Ca2+]i was performed using Fura-2, AM (Molecular Probes, Eugene, OR, USA). The cells were grown in a cover glass bottom dishes and loaded with 1 µM of Fura-2, AM, and incubated for 30 min at 37℃. The Fura-2 fluorescence was measured at a 510 nm emission with a 340/380 nm dual excitation using a PTi Sutter illuminator. Images were acquired on the cooled 3 MHz (14 bit) CCD camera (CoolSNAP HQ2; Photometrics, Tucson, AZ, USA) with 1 × 1, 2 × 2, or 3 × 3 binning under the control of MetaMorph 7.6 software. Each images were exposed over period of 100 ms.
Western blotting analyses
The 0.5–2 µg/well of cDNA was transfected into cells. After transfection for 24 h, the cells were harvested as follows. Lysates were prepared in lysis buffer (0.5% Triton X-100, 50 Tris-Cl, 150 NaCl, 1 EDTA, pH 7.5, [in mM]) by being passed through a 26-gauge needle seven to ten times after sonication. Lysates were centrifuged at 13,000 × g for 10 min at 4℃, and the protein concentration in the supernatants was determined. The proteins extracted in sample buffer were loaded onto 8% Tris-glycine SDS-PAGE gels and then subsequently transferred onto a polyvinylidene fluoride membrane.
Surface biotinylation
Phosphate buffered saline (PBS)-washed cells were incubated in 0.5 mg/ml sulfo-NHS-LC-biotin (Pierce, Rockford, IL, USA) in PBS for 30 min on ice. Afterward, unreacted biotin was quenched by the addition of 100 mM glycine in PBS. The cells were then processed as described above to prepare cell extracts. Forty microliters of a 1 : 1 slurry of immobilized avidin beads (Pierce) were added to 300 µl of the cell lysate containing 500 µg protein. After incubation for 1 h at room temperature, the beads were washed three times with 0.5% Triton-X-100 in PBS, and proteins were extracted in sample buffer. Collected proteins were then analyzed by Western blot. The experiment was performed as previously described [20].
Statistical analysis
Data were analyzed using SPSS software (IBM SPSS Statistics 23; IBM Co., Armonk, NY, USA). Results are given as mean ± standard error of the mean (SEM). Error bars indicate SEM. Here, *p < 0.05, **p < 0.01, and ***p < 0.001 were considered statistically significant, while n.s. indicates not statistically significant. Results were compared Student's t-test. All data were generated from cells pooled from at least two biologically independent experiments. No samples were excluded.
RESULTS
PLCβ downstream signals contribute to TRPC1α/4β and TRPC1α/5 inactivation
The TRPC1 subunit is not functional alone, but forms a heteromeric channel with TRPC4 or TRPC5. As a regulator, TRPC1 changes physiological properties. The current-voltage relationship of heteromeric channels shows an outward-rectifying shape, which is different from the double-rectifying shape of homomeric channels. Previously, we suggested that the Gαq protein directly interacts with TRPC4 or TRPC5 heteromeric channels and activates heteromeric channels. After activation, breakdown of PI(4,5)P2 by the Gαq-PLCβ pathway inactivates heterotetramers [11]. While studying the inactivation mechanisms of heteromeric channels, we noticed that constitutively active Gαq (Q209L) completely reduced the Englerin A (EA)-induced current to zero. EA is an agonist of TRPC4 and TRPC5 channels [21]. On the other hand, inositol polyphosphate 5-phosphatase (Inp54p) of the rapamycin-inducible system and Danio rerio voltage-sensitive lipid phosphatase (DrVSP) only partially reduced current. In other words, the channel currents were not inactivated to the zero level with maximum activation of these systems, which completely depletes the membrane PI(4,5)P2. The physiological Gαq-PLCβ pathway and these artificial lipid phosphatases hydrolyze PI(4,5) P2 differently (Fig. 1A). In the Gαq-PLCβ pathway, the guanosine triphosphate (GTP)-bound Gαq subunit activates PLCβ, which in turn hydrolyzes PI(4,5)P2 to DAG and IP3 (Fig. 1A; right). DAG acts as a second messenger that activates PKC, and IP3 releases Ca2+ from the ER. By contrast, activated DrVSP and Inp54p convert PI(4,5)P2 to phosphatidylinositol 4-phosphate (PI(4)P) and inorganic phosphate (Fig. 1A; left) [2223]. Likewise, the physiological pathway and artificial PI(4,5)P2 depletion system shows the difference in products after stimulation. We hypothesized that, if the Gαq-PLCβ downstream products are involved in an inactivation process, these signals would cause the additional channel inhibition.
Inhibitory role of IP3-cytosolic calcium on TRPC1α/4β
We first investigated the effect of IP3-induced ER Ca2+ release on TRPC1α/4β and TRPC1α/5, considering that the level of cellular Ca2+ regulates TRPC4 and TRPC5 homomeric channels [124]. The cells co-expressed channel subunits and muscarinic receptor 3 (M3R), and the calcium changes were monitored by Fura-2 calcium imaging. Application of a muscarinic receptor agonist, carbachol (CCh), activated the Gαq-PLCβ pathway and markedly increased the intracellular free calcium ([Ca2+]i) in cells expressing heteromeric channels (Supplementary Fig. 1A, B). On the other hand, there was no current inhibition in TRPC1α/4β or TRPC1α/5 when [Ca2+]i was increased by an ER Ca2+ pump inhibitor, thapsigargin (TG) (Fig. 1B–D). We measured the [Ca2+]i change in the cells expressing channel subunits and stimulated by EA (Supplementary Fig. 1C, D). The calcium change slightly increased compared to CCh stimulation. We also measured [Ca2+]i change with the rapamycin-inducible system when Gαq (Q209L, L254A) was expressed (Supplementary Fig. 1E, F). This mutant is GTPase deficient, which is constitutively active and impaired binding to PLCβ [25]. The calcium change was slightly increased compared to CCh stimulation.
To determine the [Ca2+]i function on heteromeric channels, we buffered free Ca2+ levels in pipette solutions by a Ca2+ chelator, BAPTA (Fig. 2). HEK293 cells were co-transfected with channel subunits and M3R, and the channels were stimulated by CCh. We recorded channel current changes in the whole-cell recording mode. Voltage ramp pulse from +100 mV to −100 mV over a period of 500 ms was imposed every 10 sec with a −60 mV holding membrane potential. With 0.5 mM of BAPTA, either the basal or CCh-evoked current increased in TRPC1α/4β-expressing cells (Fig. 1E). Furthermore, the currents were sustained longer than control in the presence of 0.5 mM of BAPTA. Strong titration of [Ca2+]i with 5 mM of BAPTA remarkably inhibited the current amplitude activated by CCh in TRPC1α/4β-expressing cells (Fig. 1E), whereas, in cells expressing TRPC1α/5, calcium buffering with 0.5 mM of BAPTA showed similar current changes to control, 0 mM of BAPTA (Fig. 1G). Calcium buffering with 5 mM of BAPTA also did not enhance channel currents. We normalized the trace to identify the tendency of currents change depending on time. Fig. 1F and H show the normalized heteromeric currents for pipette solution containing 0 or 0.5 mM BAPTA. The normalized currents clearly shows the effect of BAPTA on the inactivation time course of heteromeric channels. The TRPC1α/4β currents were sustained longer in the presence of BAPTA (Fig. 1F), whereas the TRPC1α/5 currents were not (Fig. 1H). This is in line with the finding that TRPC4 channels were silent when the pipette solution contained a high EGTA concentration [1]. Collectively, the results of cytosolic calcium change support the hypothesis that calcium tonically inhibits TRPC1α/4β, but not TRPC1α/5.
To investigate the additional role of Ca2+ on TRPC1α/4β current inactivation, we depleted PI(4,5)P2 first and then subsequently increased calcium. Increase of cytosolic calcium was induced by TG, a SERCA inhibitor, or ionomycin (iono) ionophore. The cells co-expressed TRPC subunits, Inp54p, and Lyn11. Channel currents were first activated by EA, and depletion of PI(4,5)P2 was induced by Inp54p with the rapamycin-induced dimerization system. As shown in Supplementary Fig. 1C and D, EA increased intracellular Ca2+ slightly. Application of TG (1 µM) completely reduced current amplitude to the basal level and showed an outward rectifying I–V shape (Fig. 2A, B). Similar current inhibition was observed in cells stimulated by ionomycin (1 µM), which raises the intracellular calcium level by allowing calcium to cross biological membranes (Fig. 2C, D). In contrast, neither type of high calcium stimulation was sufficient to inactivate the TRPC1α/5 (Fig. 2E–H). These results suggest that Ca2+ inactivates the TRPC1α/4β heteromeric channel via the Gαq-PLCβ-IP3 pathway.
Inhibitory role of DAG-PKC on TRPC1α/5
In our previous study [11], CCh stimulation induced the production of DAG and Ca2+. Since the inactivation of TRPC5 occurs via PKC-mediated phosphorylation [24], we examined the effect of the PLCβ-DAG-PKC pathway on heteromeric channels. To identify the effect of PKC on heteromeric channels, we stimulated channels by CCh (100 µM) repetitively with or without pretreatment of a PKC inhibitor, GF109203X, or chelerythrine. HEK293 cells were transfected with M3R, TRPC1, and TRPC4 or TRPC5. To quantify the inactivation, we defined the change ratio of the current at the beginning of CCh application to the currents at the end of CCh application (2 / 1 and 2′ / 1′) as the inactivation ratio (Fig. 3). As a control, CCh was applied repetitively without PKC inhibitors (Supplementary Fig. 2A, B). Both control experiments showed transient current activation by the Gαq-PLCβ pathway stimulation. The second peak current amplitude evoked by CCh stimulation was not as much as the first peak because the PI(4,5)P2 resynthesis was insufficient (Supplementary Fig. 2C, D). There was no difference between the first application of CCh and the second application of CCh (Fig. 3C; control panel). With GF109203X, TRPC1α/4β showed a similar tendency of current inactivation as the first CCh stimulation (Fig. 3A), whereas, TRPC1α/5 currents that increased during the second CCh stimulation were reversed completely (Fig. 3B, C, GF109203X panel). Similar results were observed with chelerythrine pretreatment: the inactivation ratio (2 / 1 and 2′ / 1′) was only significantly increased in TRPC1α/5 (Fig. 3C; chelerythrine panel).
Next, we used a PKC-insensitive mutant of channels, TRPC4 (T887A) and TRPC5 (T972A) for further confirmation (Fig. 4) [24]. This mutation did not affect the expression and membrane localization (Supplementary Figs. 3, 4). In the cells transfected with M3R and heteromeric channels together, we stimulated channels by CCh. Activity change of channel mutants corroborates the previous results. TRPC1α/4β (T887A) showed a similar level of enhancement to that of wild-type (Fig. 3D, E). In contrast, the TRPC1α/5 (T972A) mutant showed a large enhancement in both basal and peak current compared to wildtype TRPC1α/5 (Fig. 3G, H). To identify the tendency of current change depending on time, we normalized the current trace. We also noticed that the inactivation was prolonged in TRPC1α/5 (T972A) (Fig. 3I), but not in TRPCα/4β (T887A) (Fig. 3F). These results suggest the possibility of other downstream molecules of the Gαq pathway, especially PKC involvement in TRPC1α/5 channel regulation.
Therefore, we also investigated whether the channel responds to direct PKC activation. To induce PI(4,5)P2 depletion before PKC activation, we used a rapamycin-inducible system. HEK293 cells were transfected with channel subunits, Inp54p and Lyn11. First, we stimulated heteromers with channel agonist, EA. After that, we artificially depleted PI(4,5)P2 by translocating inp54p to membranes with rapamycin. Then, DAG analog, 1-oleoyl-2-acetyl-sn-glycerol (OAG) was added as a PKC activator. As expected, there was no further current inhibition in TRPC1α/4β after OAG stimulation (Fig. 4A, B), whereas the current amplitude of TRPC1α/5 decreased to the basal level (Fig. 4C, D). All heteromeric channels showed outward rectifying I–V curve with every drug treatment (Fig. 4A, C; left). Collectively, these results suggest that PKC via the PLCβ-DAG pathway inactivates the TRPC1α/5 heteromeric channel.
DISCUSSION
In the present study, we suggest the reinforcing inhibition of TRPC1α/4β and TRPC1α/5 channels via the Gαq-PLCβ downstream. We demonstrate that: 1) The products of PI(4,5)P2 hydrolysis, IP3 high-calcium, inactivated TRPC1α/4β. 2) On the other hand, TRPC1α/5 inactivation was under the control of the DAG-PKC level. Even TRPC4 is a close homolog of TRPC5. TRPC1/4 heteromeric channels have different inactivation mechanisms from TRPC1/5 heteromeric channels.
Forming heteromers with TRPC1 was closely related with regulation of cytosolic calcium level [2627]. Coincidentally, TRPC1 changes the permeability of homomeric TRPC4 and TRPC5 channels with decreased inward and increased outward current. This regulatory effect reduces cellular excitability. In a precedent study, knockdown of the TRPC1 channel increased the formation of TRPC5 homotetramer which in turn permeates high levels of Ca2+ and resultantly stimulates Ca2+-dependent apoptosis of neural cells in Huntington's-disease [19]. Here, our calcium data showed that calcium level change via ER release or Ca2+ influx through plasma membrane have complicated effects on heteromeric TRPC1/4 and TRPC1/5 channels. The channels are not store-operated channels considering PLCβ activation [11], and ER calcium release did not induce any changes (Fig. 1B–D). Calcium requirements in inactivation were confined to TRPC1/4, implicating that its close homolog, TRPC5, has a different regulation mechanism. Even calcium acts as a second messenger, both hetero channels could not be activated by CCh stimulation with 5 mM BAPTA in pipette solution (Fig. 1E, G). Therefore, further study is needed to understand the exact calcium concentration the regulation of heteromeric channel activities depend on.
All mammalian TRPCs are regulated by downstream of PLC [28]. TRPCs responds to PLC activation in an analogous way. Other grouped TRPC channels, TRPC3, 6, and 7, required both PI(4,5)P2 and its downstream product, DAG, to maintain the channel activities [45]. TRPC4 and TRPC5 homotetrameric channels were also maintained by PI(4,5)P2 [3] but inhibited by active PKC binding to DAG [29]. Initially, it has been proposed that application of DAG itself had no response in TRPC5 [30]. Recent results have shown that the C-terminal PDZ-binding motif of TRPC4 and TRPC5 regulates DAG-sensitivity [31]. PKC inhibitor or TRPC5 (T972A) mutant conferred DAG-sensitivity to TRPC4 and TRPC5 channels [31]. In our attempts, PKC also inactivated TRPC1/5 but not TRPC1/4 channels (Fig. 4). Although Storch et al . [31] showed that PI(4,5)P2 depletion also conferred DAG-sensitivity to TRPC4, 5 channels, OAG decreased the heteromeric TRPC1/5 current in our study via PI(4,5)P2 depletion (Fig. 4).
Low cytosolic calcium levels significantly increased current amplitude of TRPC1/4 (Fig. 1D), while PKC-insensitive mutant showed significantly increased current amplitude of TRPC1/5 (Fig. 3F). Myeong et al. [11] also showed that U73122, an inhibitor of PLCβ, increased the basal current in HEK293 cells expressing heteromeric channels. These results suggest that there is endogenous PLC activity in HEK293 cells expressing heteromeric channels. The inactivation of CCh-induced current under 0.5 mM of BAPTA in TRPC1/4 (Fig. 1D) represent the inactivation due to PI(4,5)P2 depletion and remains at the higher level compared with that under 0 mM of BAPTA. Similar results obtained in PKC-insensitive mutant TRPC1α/5 (T972A) (Supplementary Fig. 3). These results also suggest that Ca2+ and PKC are involved in the inactivation process of heteromers as well as PI(4,5)P2 depletion.
PLC modulation on TRPCs is inseparable with PI(4,5)P2, the substrate of PLC. TRPC1/4 and TRPC1/5 channels interact with the PH domain of PLCδ through PI(4,5)P2 [32]. PI(4,5)P2 was shown to be essential for TRPC4 activation [3]. Additionally, the cytosolic calcium level, calmodulin, and PKC tightly control TRPC4 and TRPC5 function [116171824]. It has been proposed that TRPC4-Gαi/o protein activation depends on PLCδ1 [1]. They also suggested that PLCδ1 would play a central role in TRPC4 regulation with PI(4,5)P2-Ca2+ level. In contrast to other isozymes, calcium ion concentration is the main regulator of PLCδ1 activity. Activated PLCδ1 by high calcium concentration hydrolyzes membrane PI(4,5)P2, which is essential for channel activity. Actually, EA stimulation and rapamycin-inducible system of Gαq (Q209L, L254A) did not increase the cytosol calcium level compared to CCh stimulation (Supplementary Fig. 1), which explains the maintenance of heterotetrameric channel activity. In a preceding study, TRPC4 homotetramer activated by EA significantly fluctuated compared to heteromeric TRPC1/4 channels [26]. High calcium influx though TRPC4 channels activate PLCδ more strongly; hence, PI(4,5)P2 hydrolysis and channel inhibition was facilitated. The PLCδ isoform may serve as a key channel function regulator by regulating second messengers. Therefore, the mechanism by which the PLCδ-Ca2+ relationship affects TRPCs tetrameric channels must be considered.
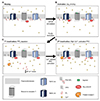
Fig. 5 illustrates a model for the TRPC1/4 and TRPC1/5 channel regulation in Gαq-PLCβ downstream events. Overall, our results provide an important feedback loop mediated by Ca2+ and DAG. The TRPC1/4 and TRPC1/5 heteromeric channels are regulated by the Gαq-PLCβ pathway as follows: A) In the resting state, PI(4,5)P2 is directly bound to TRPC1/4 or TRPC1/5, and maintains heteromeric channels. Guanosine diphosphate (GDP) bound inactive Gαq has been shown to be pre-coupled with Gαq-coupled receptors physically. B) Upon receptor activation with specific ligand, in turn, Gαq-coupled receptors activate the Gαq by exchanging a molecule of GDP to GTP. At this point, GTP-bound Gαq releases from the receptor and directly interacts with heteromeric channels. This interaction induces the channel activation state, allowing calcium to diffuse inside the cells. C) PLCβ activation accelerates cleavage of membrane-bound PI(4,5) P2 into the IP3 and DAG. The first inactivation of heteromeric channels happens through decreasing membrane PI(4,5)P2 levels. D) Following the first inactivation, IP3 diffuses to the cytosol and induces calcium release from the ER. This causes intracellular calcium increase, which leads to the second phase of inactivation specific to TRPC1/4. The DAG remains in membrane and acts as a second messenger that activates PKC. The function of calcium-bound PKC is closely coupled with the TRPC1/5 inactivation process in a DAG-indirect manner. Collectively, the strong inactivation mechanism in the Gαq-PLCβ pathway is distinct for TRPC1/4 and TRPC1/5 heteromers.




 XML Download
XML Download