

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Omega-3 fatty acids are known to have anti-inflammatory, anti-thrombotic, anti-arrhythmic, hypolipidemic and vasodilatory properties [1]. Docosahexaenoic acid (DHA), an omega-3 fatty acid, was shown to have beneficial effects in critically ill patients, especially under conditions of impaired cardiovascular health, e.g., coronary heart disease [2], or deteriorated pulmonary function, e.g., acute respiratory distress syndrome or acute inflammatory pulmonary disease [3]. These beneficial effects associated with DHA treatment may be related to endothelial cell function [4]. Endothelial cells play important roles in regulation of vessel tone, vessel permeability, thrombosis and leukocyte recruitment, which are controlled through endothelial cell calcium signalling and/or modulation of nitric oxide (NO) production. However, previous studies addressing the effects of DHA in the cardiovascular system rather focused on cardiomyocytes and vascular smooth muscle cells, while little information is available for endothelial cells. In rat cardiomyocytes acute application of DHA reduced the doxorubicin-induced calcium increase via inhibition of sarcoplasmic reticulum calcium release [5]. In this cell model, DHA eliminated the cytosolic calcium increase induced by endothelin-1 [6]. In rat vascular smooth muscle cells DHA blocked the cytosolic calcium transient induced by 5-hydroxytryptamine [7]. In 2001, Kuroda et al. [8] showed in bovine aortic endothelial cells that circulating unsaturated fatty acids (linoleic acid and oleic acid) inhibited an adenosine triphosphate (ATP)-γS-induced calcium increase. The same effect was observed when human umbilical vein endothelial cell (HUVEC) were treated with unsaturated fatty acids using histamine as the stimulus for calcium elevation. Unfortunately, in these studies on endothelial cells the omega-3 fatty acid DHA was not included, leaving it to be tested whether DHA blunts the agonist-induced calcium transient in endothelial cells. More recently, Ye et al. [9] published that DHA suppressed oxidative stress-induced calcium influx in the human endothelial cell line EAhy926 by displacement of TRPC1–the component of calcium entry channels–from membrane caveolar lipid rafts. However, the variability of the results obtained in different cell species and the limited data obtained in endothelial cell studies undertaken so far do not reveal a comprehensive figure of the regulation of the calcium concentration in endothelial cells in response to DHA treatment.
Endothelial cells are known to release ATP under various conditions e.g. shear stress and hypoxia-reperfusion [1011]. Once released, extracellular ATP enhances in an autocrine manner the endothelial cytosolic calcium and may subsequently increase endothelial cell nitric oxide production. As shown before, release and extracellular metabolism of ATP in endothelial cells are modulated by DHA resulting in an enhanced extracellular ATP concentration [12]. Thus, hypothetically a causal link may exist between the modulation of membrane lipid composition by DHA, extracellular ATP, endothelial cell calcium regulation and the generation of nitric oxide.
It is well documented that ATP elevates both the cytosolic calcium level and NO production [13]. Extracellular ATP released from endothelial cells by enhanced shear stress acts by binding to G-protein coupled P2-receptors [1415]. ATP stimulation of endothelial cells was shown to activate the IP3 pathway [16] thereby augmenting the cytosolic calcium concentration which after interacting with calmodulin may result in endothelial cell NO synthase (eNOS) activation. Alternatively, ATP can also activate eNOS via phosphorylation through various kinases in a calcium-independent manner [14]. Whether DHA may interfere with eNOS phosphorylation which may modulate eNOS activity is unclear to date.
Based on these considerations the aims of the present study were to address the impact of DHA on the ATP stimulated Ca2+-transient of endothelial cells. Furthermore, it should be clarified whether this DHA dependent effect on endothelial cell calcium was primarily dependent on transmembrane calcium influx and the potential role of store-operated calcium channels should be objectified. In addition, we wished to test whether the modulation of the calcium transient by DHA subsequently affects eNOS phosphorylation or whether DHA may change baseline eNOS phosphorylation independent of ATP stimulation.
Go to : 
METHODS
Cell culture
HUVEC were isolated and maintained in supplemented M199 (Gibco; Life Technologies, Breda, The Netherlands) in 5% CO2 atmosphere and 37℃ (Heraeus BBD 6220 incubator; Thermo Scientific, Waltham, MA, USA). The cells were split for experiments when they had reached a confluent state. Approximately 50,000 cells were seeded on a 3.5 cm cell culture dish and supplemented with culture medium ECGM-2 (Promocell, Heidelberg, Germany). When the cells were 80%–90% confluent, the medium was renewed and supplemented with antioxidants including 50 µM of alpha-tocopherol (Sigma, Deisenhofen, Germany) and 300 µM of ascorbic acid (solution prepared from the pharmacy department of the University Hospital Dresden, Germany). The next 24 h were suitable for DHA treatment. DHA (IBL, Hamburg, Germany) diluted in ethanol to a stock solution of 761 mM was and further diluted just before adding the solution to the cells in the culture medium supplemented with antioxidants at final concentrations of 0, 3, 12, 50 µM.
Intracellular calcium measurement
To permit visualization under the fluorescent microscope, the cells were cultured on glass-bottom cell culture dishes which were made from normal 3.5 cm plastic dishes. A 1 cm2 hole was cut in the centre of the dish bottom and covered by a thin glass piece (Menzel Gläser; Thermo Fisher Scientific, Braunschweig, Germany) with the thickness of 0.13 to 0.16 mm glued to the plastic material. For growth on glass dishes HUVEC required a special gelatine-coating procedure with 0.5% gelatine cross-linked with 2% glutaraldehyde and 2 mM glycine.
Intracellular calcium was measured using the cell membrane permeable fluorescent dye Fura-2-AM which was excited at two wavelengths 340 nm (calcium free) and 380 nm (calcium bound). Emission was determined at 510 nm. HUVEC were loaded with a mixture of 5 µM Fura-2-AM (Invitrogen, Carlsbad, CA, USA) and 0.5% Fluronic F127 (Invitrogen) for 20 min at room temperature. After this a further 20 min reaction time at 37℃ was allowed in order to achieve hydrolysation of the acetoxymethyl groups by endogenous esterases to generate the fluorescent form of the dye. After Fura loading, depending on the aims of the experiments, further inhibitors or substances were added as detailed below. To start the calcium recording, the cells were stimulated with 100 µM ATP (Sigma) at 37℃. Fluorescence intensity was detected by fluorescence microscopy (Axiovert 100M; Zeiss, Thornwood, NY, USA) equipped with an Axiocam MR rev3 camera (Zeiss) using a time frame of 1 sec, an exposure time of 40 ms for 340 nm and 15 ms for 380 nm excitation wavelength, respectively. The results were processed by a calcium imaging software Axiovision Release 4.8 provided by Zeiss (2009). After subtraction of the background fluorescence the ratio between F340/F380 signals represented the change of the intracellular calcium level. All calcium signals were recorded at exactly 37℃.
Western blot
Western blot was performed with gels from 10% to 12% polyacrylamide depending on the size of the target protein. Based on the sample protein concentration the appropriate volume for sample dilution was calculated in order to give equal total protein amounts (20 to 30 µg total protein/lane). After sample loading, electrophoresis on SDS-PAGE was performed at 80 V for the stacking gel and at 120 V for the resolving gel in running buffer to separate the proteins according to their molecular weight. In the next step, proteins on the gel were transferred onto nitrocellulose membrane following the kit instruction with semi-dry blotting solution at 185 mA for 90 min. The membrane then was blocked with 5% skim milk in TBS-Tween 0.1% (for non-phosphorylated proteins) or with 5%BSA in TBS-Tween 0.1% (for phosphorylated proteins) at room temperature and gentle shaking for 1 h. The antibody was diluted in the blocking solution. The membrane was incubated with the primary antibody for 16 h at 4℃ or 1–2 h at room temperature. The primary antibodies in this study were mouse anti-eNOS/NOS type III (1:1,000; BD Transduction, San Jose, CA, USA), rabbit anti-P-ser1177-eNOS (1:1,000; Cell Signalling, Beverly, MA, USA), rabbit anti-P-thr495-eNOS (1:1,000; Cell Signalling), mouse anti-α-tubulin (1:1,000; Sigma), mouse anti-β-actin (1:1,000; Sigma). The membrane was washed three times at room temperature, 10 min each with TBS-Tween 0.05%. A suitable secondary antibody used to detect the first antibody was added to the membrane for 60 min at room temperature with gentle shaking. We used goat anti-mouse (1:1,000; Dianova, Hamburg, Germany) and goat anti-rabbit (1:1,000; Dianova or BD Transduction) as secondary antibodies. The membrane was washed three times at room temperature, for 10 min each, with TBS-Tween 0.05% and developed with a chemi-luminescent reagent before detection. An Alias 3000 Reader was used to visualize the protein band on the membrane with appropriate exposure time and resolution.
The blot images were quantified densitometrically using ImageJ software (National Institutes of Health; https://imagej.nih.gov/ij/). The target protein amounts were normalized to the tubulin or the actin protein which were taken to represent housekeeping proteins in each sample. The protein ratios were normalized to the control of each experiment.
Statistical analysis
Statistical analysis was performed using SPSS for Windows version 15.0. The results were expressed as means ± standard error. Differences between mean values of two groups were analyzed by Student's t-test. When more than two groups were compared, statistical analysis was performed by a one-way ANOVA followed by post-hoc test. A p-value < 0.05 was considered to indicate a statistically significant difference.
Go to :
RESULTS
Dose-response effect of DHA on ATP-induced intracellular calcium concentration
As expected application of ATP (100 µM) elevated the intracellular calcium concentration (Fig. 1A–C). The maximum effect was equivalent to an increase of the ratio 340/380 from 0.8 ± 0.02 to 2.5 ± 0.04 (p < 0.001, n ≥ 5; Fig. 1B). Preincubation with DHA for 48 h dose-dependently decreased the calcium transient evoked by ATP in HUVEC equivalent to a decrease of the 340/380 ratio to 2.16 ± 0.03, 2.05 ± 0.03 and 2.05 ± 0.03 at DHA concentrations of 3, 12 and 50 µM, respectively (p < 0.001, n = 4; Fig. 1B). The inhibitory effect of DHA (12 µM) on the ATP-induced calcium signal affected the entire Ca2+-transient as exemplified in Fig. 1C. This DHA concentration was chosen for all consecutive experiments.
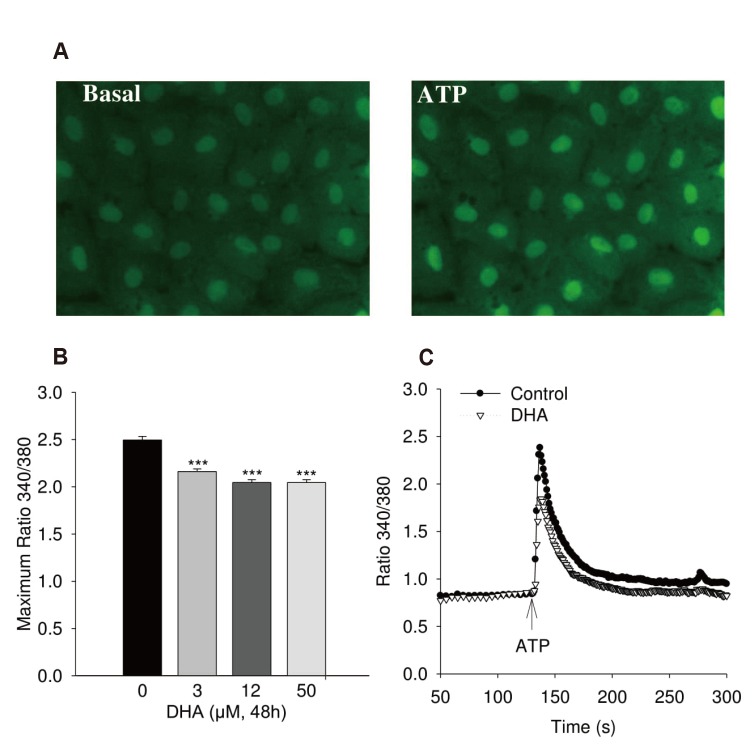 | Fig. 1Calcium increase of human umbilical vein endothelial cell (HUVEC) induced by adenosine triphosphate (ATP).(A) Typical fluorescence image. Typical fluorescence of Fura-2AM loading cell image with 40× magnification. (B) Maximum of ATP (100 µM) induced calcium transient in HUVEC treated with 0, 3, 12, 50 µmol/l docosahexaenoic acid (DHA) (n = 4). (C) Kinetic of calcium transient in cells stimulated with ATP (100 µM) without and with treatment with 12 µM DHA for 48 h (n = 8). Cells were loaded with 5 µM Fura-2-AM. ANOVA; ***p < 0.0001 between DHA treatment and control.
|
Effect of DHA on ATP-induced eNOS phosphorylation
Neither DHA treatment nor ATP stimulation nor the combination of both had any effect on total eNOS protein expression (Fig. 2A, B). Because eNOS is activated when phosphorylated at residue ser1177 and in-activated when phosphorylated at residue thr495 [17] we tested the impact of ATP and DHA on eNOS phosphorylation.
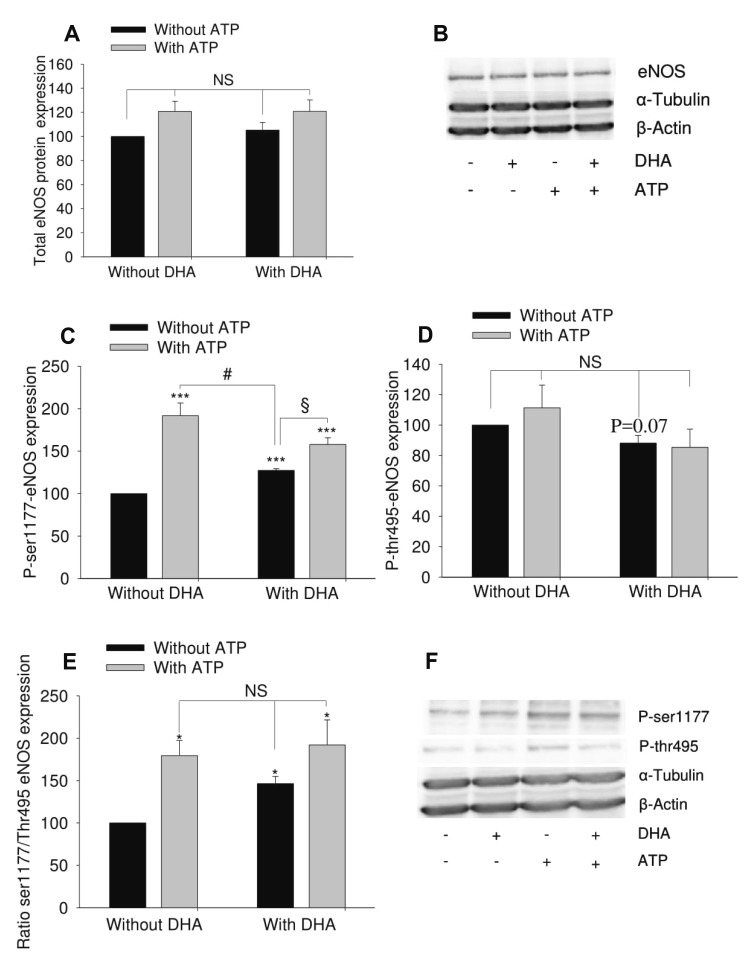 | Fig. 2Effect of docosahexaenoic acid (DHA) on adenosine triphosphate (ATP)-induced endothelial nitric oxide synthase (eNOS) phosphorylation in human umbilical vein endothelial cell (HUVEC).HUVEC were treated with 12 µM DHA for 48 h. 100 µM ATP was used to stimulate eNOS for 1 min. (A) Total eNOS expression (n = 7). (B) Representative blot. (C) Phosphorylated eNOS at ser1177 residue (n = 5). (D) Phosphorylated eNOS at thr495 residue (n = 5). (E) Ratio of phosphorylated eNOS at ser1177 and phosphorylated eNOS at thr495 (n = 5). (F) Representative Western blot. The results show mean ± standard error of densitometric quantification of blots. ANOVA was used to test for differences. *p < 0.05, ***p < 0.001 vs. untreated group (no DHA, no ATP); #p < 0.05, §p < 0.05. NS, no significant difference.
|
As seen in Fig. 2C, ATP induced a strong rise of eNOS phosphorylation at ser1177 without and with 48 h exposure to DHA. Also, DHA (12 µM) elevated ser1177 phosphorylation of eNOS to 127.3 ± 2.1%. In contrast to the strong change of eNOS phosphorylation at ser1177, ATP treatment did not significantly change eNOS phosphorylation at thr495. Also, treatment with DHA did not significantly change eNOS phosphorylation at thr495 residue (although there was a trend toward a decrease, p = 0.07; Fig. 2D). The ratio of eNOS phosphorylation at ser1177 versus thr495 is shown in Fig. 2E. Compared to control conditions, ATP enhanced this ratio to 179.3 ± 18% and 192 ± 29.7% in groups untreated and treated with DHA, respectively. It is also evident that DHA treatment itself increased the ser1177/thr495 ratio to 146.4 ± 8.6%. This suggested that DHA caused activation of eNOS. Representative western blots are shown in Fig. 2F.
Effect of DHA on ATP-induced calcium increase depends on extracellular calcium
Chelation of intracellular calcium with BAPTA (10 µM, 20 µM) lowered the intracellular calcium signal under baseline conditions (not shown) and largely changed the calcium rise after stimulation with ATP (Fig. 3A, B).
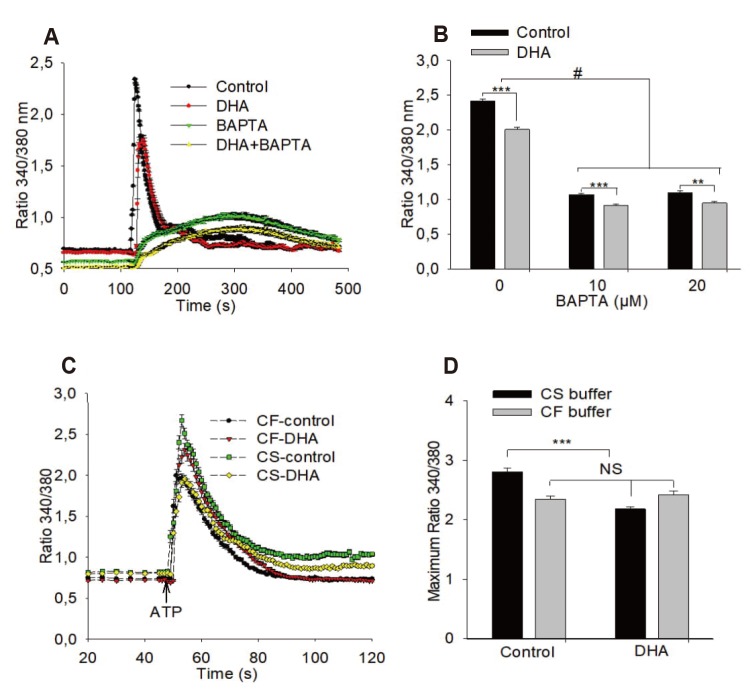 | Fig. 3Effects of BAPTA and withdrawal of extracellular calcium on adenosine triphosphate (ATP) induced calcium transient.(A, B) Presence of BAPTA (10 µM, 20 µM) does not eliminate the effect of docosahexaenoic acid (DHA) on ATP-induced calcium transient (n = 3). (C, D) Elimination of extracellular calcium reduces the ATP-induced calcium transient and abolishes the effect of DHA on the calcium transient (n = 4). DHA 12 µM, ATP 100 µM; ANOVA/Tukey's post-hoc test was used: **p < 0.001; ***p < 0.0001, #p < 0.0001 between groups as indicated by brackets,. NS, no significant difference; CS, calcium-saturated; CF, calcium-free. For further information see methods.
|
This effect was seen in cells untreated as well as treated with DHA for 48 h. However, BAPTA treatment did not abolish the inhibitory effect of DHA on ATP-induced calcium increase (difference DHA versus respective control in Fig. 3A, B). We also studied the effects of calcium removal from the cell buffer (Fig. 3C, D). Here HUVEC were studied in a calcium-free buffer (0 mM) and a calcium-saturated buffer (1.8 mM), respectively. Under baseline conditions, the calcium-free buffer caused a small decrease of the intracellular calcium content in comparison with the calciumsaturated buffer (not shown). The ATP-induced calcium increase was significantly blunted in absence of extracellular calcium indicating that extracellular calcium played a role for the cytosolic calcium increase evoked by ATP. Importantly, the inhibitory effect of DHA on the increase of cellular calcium was abolished in calcium free buffer. This indicated that DHA treatment affected the calcium influx in human endothelial cells.
DHA inhibits ATP-induced calcium influx via store-operated calcium channels that activate eNOS
To test whether store-operated calcium channels were involved in the ATP stimulated calcium transient, 2-aminoethoxydiphenyl borate (2-APB), a blocker of store-operated calcium channels, was used. Two sets of experiments were performed, one without DHA treatment, the other with DHA treatment (12 µM) for 48 h. After loading cells with Fura-2-AM, incubation with 75 µM 2-APB started for 30 min. The maximum calcium increase after ATP stimulation is shown in Fig. 4A, B. In cells untreated with DHA, 2-APB treatment significantly decreased the ATP-induced calcium signal from 2.58 ± 0.05 to 1.94 ± 0.04. Without the inhibitor 2-APB, DHA reduced the ATP-induced calcium increase (Fig. 4A, B) as reported above (Figs. 1C, D and 3A, B). However, when cells had been treated with DHA the presence/absence of 2-APB had no effect on the 340/380 nm ratio after ATP stimulation. This indicated that the effect of DHA on the ATP-induced intracellular calcium increase depended on functionally active store-operated calcium channels.
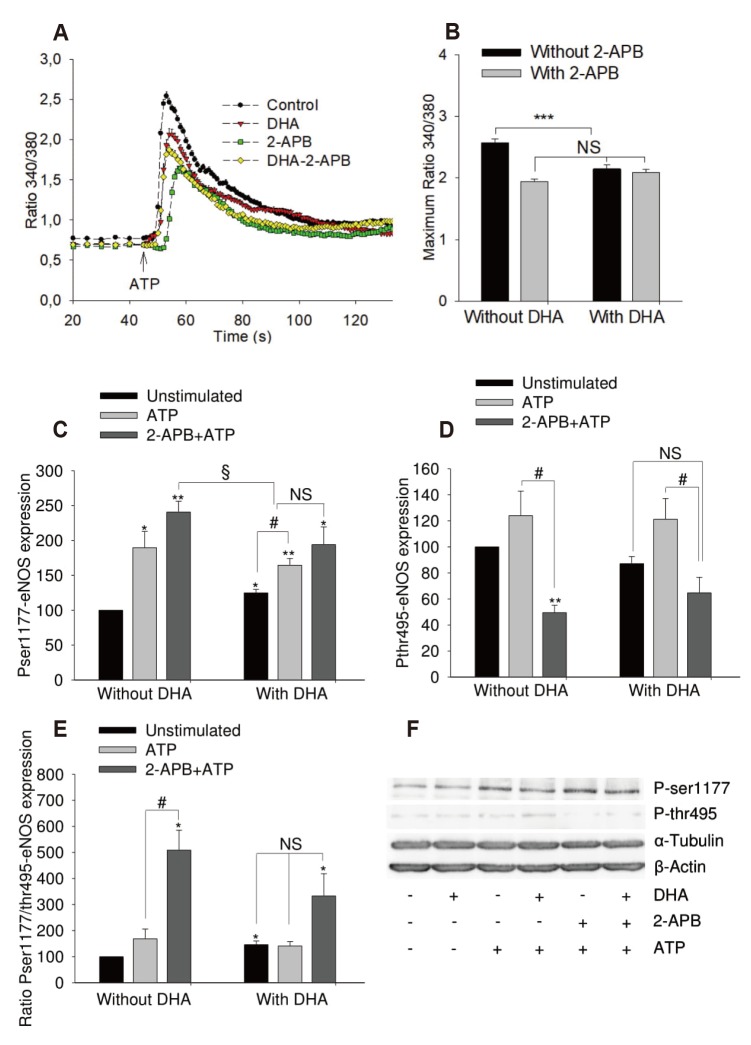 | Fig. 4Effects of 2-aminoethoxydiphenyl borate (2-APB) on adenosine triphosphate (ATP) induced calcium transient and endothelial nitric oxide synthase (eNOS) phosphorylation of human umbilical vein endothelial cell.Cells were left untreated or treated with 12 µM of docosahexaenoic acid (DHA) for 48 h. (A, B) Effect of 2-APB (75 µM, 30 min at 37℃) on ATP-induced (100 µM) calcium increase. ANOVA/Tukey's post-hoc test, n = 4, ***p < 0.0001 vs. untreated group. (C–F) Effects on eNOS phosphorylation. Results show mean ± standard error of densitometric quantification of blots from 4 independent experiments. ANOVA was used to test for differences between groups. *p < 0.05, **p < 0.01 vs. untreated group; #p < 0.05, §p < 0.05. NS, no significant difference.
|
In additional experiments we addressed the question whether inhibition of store-operatedcalcium channels has an impact on the phosphorylation of eNOS. The experimental procedure was as described for data shown in Fig. 4A, B. As shown in Fig. 4C, without DHA treatment, ATP elevated ser1177 phosphorylation of eNOS by 189.6 ± 23.5%. After 2-APB treatment the ATP-induced increase of phosphorylation of eNOS at ser1177 amounted to 240.6 ± 15.6%. Similar results were obtained in cells treated with DHA. However, as noted above the extent of elevation of ser1177 phosphorylation associated with ATP treatment was diminished in presence of DHA. With respect to eNOS phosphorylation at thr495 it was found that 2-APB strongly blunted the ATP induced phosphorylation of eNOS at thr495 (48.2 ± 5.8% of control). A similar decrease of eNOS phosphorylation at thr495 by 2-ABP was seen after treatment with DHA for 48 h (Fig. 4D). Because 2-APB augmented phosphorylation at ser1177 and diminished phosphorylation at thr495, the ratio of phos-ser1177 to phosthr495 eNOS was markedly increased (Fig. 4E). Representative western blots are shown in Fig. 4F. In conclusion, the blocker of store-operatedcalcium channels, 2-APB, increased eNOS phosphorylation at ser1177 and diminished phosphorylation at thr495, hence suggesting a considerable activation of the eNOS protein.
Effect of calcium store depletion by thapsigargin on calcium signal and eNOS phosphorylation
Thapsigargin is known to increase intracellular cytosolic calcium by activating its release from calcium stores as well as by an additional transmembraneous influx. Thapsigargin inhibits the endoplasmic reticulum (ER) calcium pump resulting in inhibition of calcium uptake and hence calcium store depletion. When HUVEC were exposed to thapsigargin (2 µM), the calcium signal was largely enhanced over several minutes (Fig. 5A, B). After 450 sec, when the calcium signal was in a steady state, application of ATP induced a small Ca2+-transient, which was clearly reduced as compared to the absence of thapsigargin (Fig. 5A, B). In presence of thapsigargin the Ca2+-transient in response to ATP was not anymore diminished after DHA treatment (Fig. 5A, B). This was taken to suggest that calcium stores (ER) and potentially thereby controlled store-operated calcium channels may play a role in the cytosolic calcium increase in response to ATP.
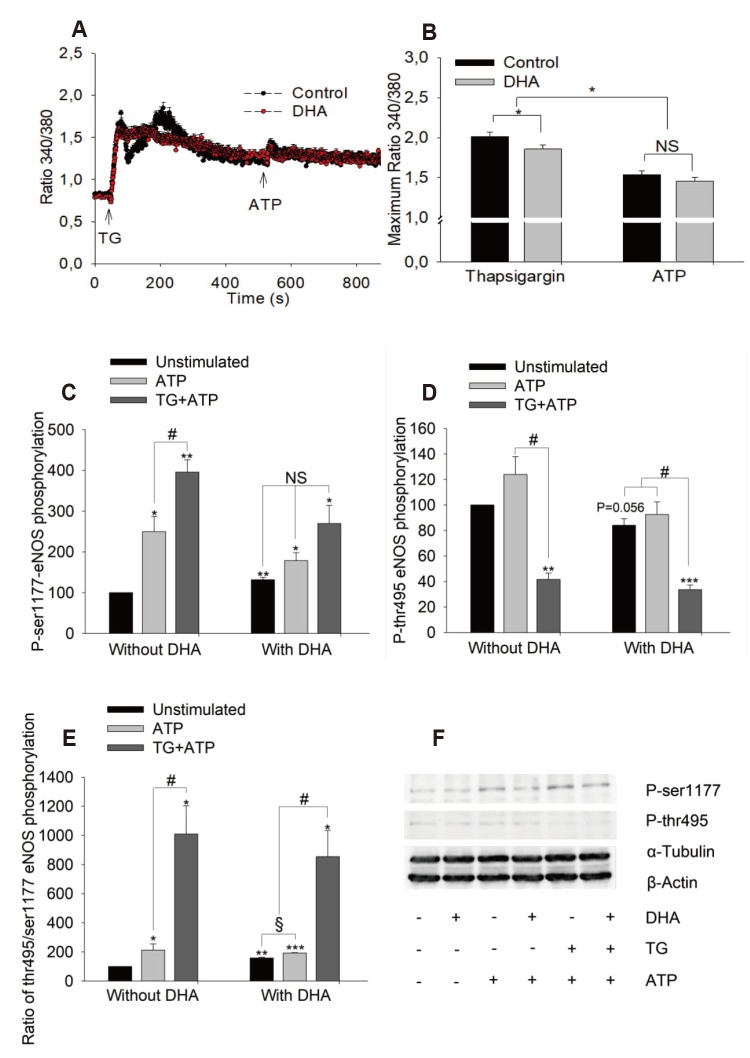 | Fig. 5Effects of calcium store-depletion by thapsigargin on adenosine triphosphate (ATP)-induced maximum calcium increase and endothelial nitric oxide synthase (eNOS) phosphorylation.(A, B) Maximum of calcium increase in presence of thapsigargin (2 µM) on the left and following ATP (100 µM) in presence of thapsigargin on the right. The calcium signal was recorded following thapsigargin (TG) for 450 sec before ATP was applied. Cells were either treated with docosahexaenoic acid (DHA) 12 µM for 48 h or left untreated (control), n = 3, 37℃. (C–F) Effects of TG on eNOS phosphorylation. Cells were stimulated with TG (1 µM, 10 min) before stimulation with ATP (100 µM, 1 min). Samples were harvested and stored at −80℃ for western blot. Results show mean ± standard error, n = 4. ANOVA/Tukey's post-hoc test was used to test for differences between groups. *p < 0.05, **p < 0.01, ***p < 0.001 vs. untreated group; #p < 0.05, §p < 0.05. NS, no significant difference.
|
To address a potential role of store-operated calcium channels on the ATP-induced phosphorylation of eNOS in HUVEC, the ER calcium-ATPase-pump inhibitor thapsigargin was used to deplete intracellular calcium stores. Probably, this consequently inhibited the store-operated calcium channel [18]. Under this condition ATP was applied to stimulate eNOS phosphorylation. As shown in Fig. 5C–F, a similar result was obtained as using the store-operated calcium channel blocker 2-APB (Fig. 4C–F). Thapsigargin on one hand further elevated ATP-induced phosphorylation of eNOS at ser1177 residue (Fig. 5C), while on the other hand it strongly decreased ATP-induced phosphorylation of eNOS at thr495 residue (Fig. 5D). Therefore, in presence of thapsigargin the ratio between the active form (ser1177-phosphorylated eNOS) and the inactive form (thr495-phosphorylated eNOS) in HUVEC was markedly increased. Effects were similar, both with and without DHA treatment (Fig. 5E, F). Representative western blots are depicted in Fig. 5F. This result is in support of that seen with 2-APB and suggests that store-operated calcium channels play an important role in phosphorylation of eNOS at ser1177 and thr495 residues in HUVEC.
Evidence for a P2R-mediated effect after ATP stimulation
Endothelial cells express purinergic receptors including P2X (P2X4, P2X5, P2X7) and P2Y (P2Y1, P2Y2, P2Y4, P2Y12). The shear stress dependent ATP release is reported to activate endothelial P2Y2 receptors resulting in eNOS activation [14]. Here, we tested the involvement of P2-receptors in the ATP-induced calcium increase by using the non-specific P2 receptor inhibitor PPADS. Incubation of HUVEC with 100 µM PPADS for 30 min blunted the increase of the intracellular calcium signal following 100 µM ATP under baseline conditions as well as in presence of DHA (Fig. 6A, B). Thus, the ATP-stimulated calcium increase depended strongly on activation of P2-receptors. However, the inhibiting effect of DHA on ATP-induced calcium increase is independent of P2R activation.
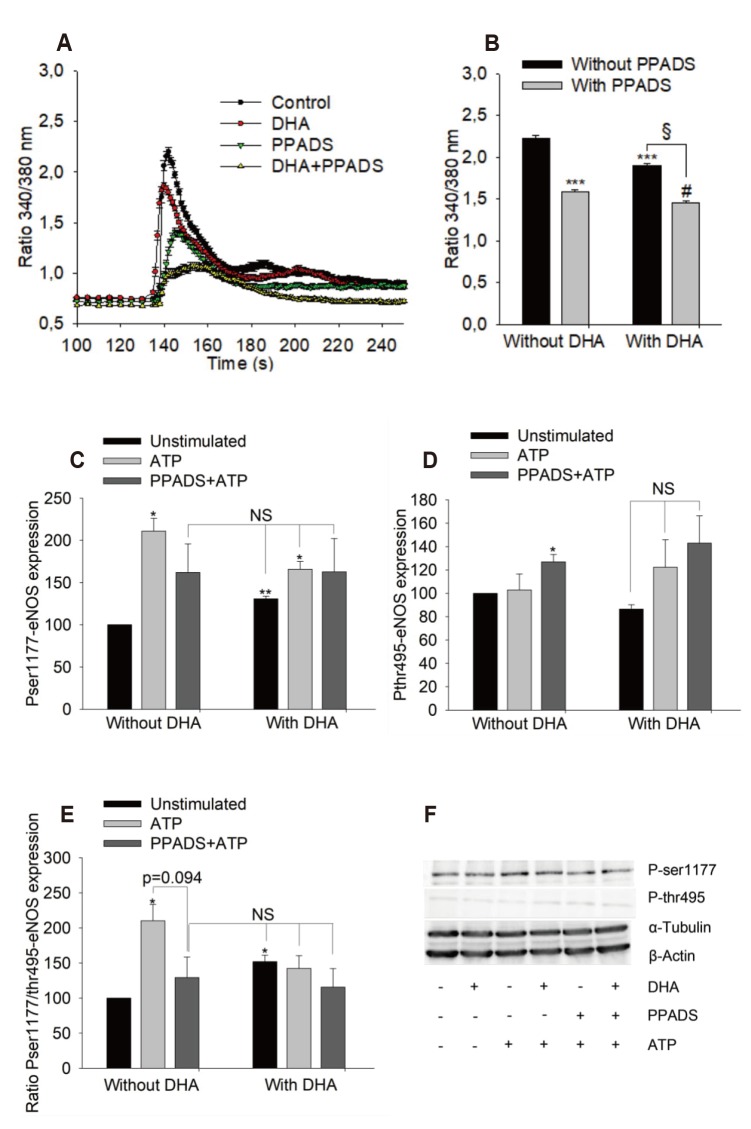 | Fig. 6Effects of PPADS on adenosine triphosphate (ATP)-induced maximum calcium signal and endothelial nitric oxide synthase (eNOS) phosphorylation.(A, B) Maximum calcium signal in human umbilical vein endothelial cell treated with 12 µM docosahexaenoic acid (DHA) for 48 h or left untreated (control), n = 3, PPADS 100 µM for 30 min at 37℃. (C–F) Effects of PPADS on eNOS phosphorylation. Results show mean ± standard error, n = 3. Data was tested using ANOVA/Tukey's post-hoc test to compare different groups. *p < 0.05, **p < 0.01, ***p < 0.0001 vs. untreated group; §p < 0.0001 between DHA treated groups with and without PPADS; #p < 0.01 between PPADS treated groups with and without DHA. NS, no significant difference.
|
In a further set of experiments the effects of the P2R blocker PPADS on the ATP-induced change of eNOS phosphorylation was assessed in HUVEC untreated or treated with DHA. As depicted in Fig. 6C the increase of ser1177 eNOS phosphorylation was blunted in the presence of PPADS. PPADS treatment significantly increased phos-thr495 eNOS after combined treatment with PPADS and ATP relative to ATP treatment alone (Fig. 6D). In the absence of DHA, PPADS powerfully reduced the ATP-induced increase of the ratio ser1177/thr495 phosphorylation of eNOS (Fig. 6E, F).
Go to :
DISCUSSION
The present study shows that DHA treatment of HUVEC dose-dependently reduced the ATP-induced calcium transient. This effect was preserved in the presence of BAPTA, but it was eliminated after withdrawal of extracellular calcium or addition of 2-APB or thapsigargin. In addition, DHA increased the ser1177/thr495 phosphorylation of eNOS under baseline conditions but did not affect this ratio during ATP stimulation. In the following, we will discuss these findings before conclusions are reached.
Intracellular calcium homeostasis is highly complex. Several intracellular compartments as the ER and mitochondria contribute to the regulation of the cytosolic calcium concentration. In addition, transmembrane calcium fluxes enhance the cytosolic calcium concentration and may trigger calcium release from intracellular stores [19]. Endothelial cells are known to release ATP to the extracellular environment [20]. This ATP release may result in autocrine regulation of endothelial cell function [14]. The present set of experiments revealed that application of DHA in HUVEC for 48 h resulted in an inhibition of the ATP-induced calcium transient in a dose-dependent manner (Fig. 1B, C). This led to the question whether DHA inhibited calcium influx across the cell membrane or whether it inhibited calcium release from intracellular stores. Sergeeva et al. [21] provided evidence that DHA inhibited the thrombin-induced calcium increase in rat astrocytes by liberation of endogenous arachidonic acid and consecutive inhibition of calcium release from the ER. A similar result and explanation were given for the inhibitory effect of DHA on doxorubicin-induced calcium increase in rat ventricular cardiomyocytes and tumor cell colonocytes [5]. However, others provided evidence that the inhibitory effect of DHA on the calcium transient was based on inhibition of calcium influx across the cell membrane in rat vascular smooth muscle cells [7]. Recently, in a study on oxidative stress induced calcium signalling performed in a human endothelial cell line (EAhy926), DHA inhibition of calcium influx was explained by an altering of lipid composition in membrane caveolar rafts [9]. In the present study, we used different approaches to assess the mode of action of DHA on the ATP-induced calcium transient. Using different concentrations of the calcium chelator BAPTA to eliminate the intracellular calcium, the ATP-induced calcium transient was markedly blunted in both DHA treated and untreated groups. However, BAPTA treatment did not modify the inhibitory effect of DHA on the ATP-induced calcium increase (Fig. 3A, B) suggesting that BAPTA did not interfere with the primary site of action of DHA. In contrast, incubation of endothelial cells in a calcium free buffer (Fig. 3C, D) eliminated the inhibitory effect of DHA on the ATP-induced calcium transient. These results support the view that in endothelial cells DHA primarily inhibits the ATP-induced calcium influx across the membrane. ATP is known to induce the calcium influx via different channels including P2X4 in human endothelial cells [22] and TRPV4 in rat carotid artery endothelial cells [23]. With respect to different cell species the different calcium channels may contain various subunits. For simplification, one may define two general types of membrane calcium influxes: 1) the first is dependent on calcium release from the intracellular stores called store-operated calcium influx and 2) the second is a calcium influx which occurs independently of a store-operated calcium release. Application of 2-APB, a blocker of store-operated calcium channels (Fig. 4A, B) inhibited ATP-induced calcium increase. 2-APB had no effect on the ATP-induced calcium transient in cells treated with DHA. This indicates that DHA interfered with store-operated calcium channels. Thapsigargin is a frequently used inhibitor of the calcium ATPase-pump expressed on the ER membrane. It depletes calcium from this intracellular calcium store and by inhibiting calcium accumulation an inactivation of the store-operated calcium release results [19]. Thapsigargin was used in the current experiments to confirm whether DHA exerted its inhibitory effect on the membrane calcium influx via store-operated calcium channels. Typically, thapsigargin elevated cytosolic calcium, probably resulting from a release by intracellular stores and influx from the extracellular environment in parallel with inhibition of calcium uptake into intracellular stores. Hence, the intracellular store was emptied, while the cytosolic calcium concentration increased. In DHA treated cells the thapsigargin-induced elevation of cytosolic calcium was reduced. However, when cells were stimulated afterwards with ATP, there was no difference between the DHA-untreated and DHA-treated groups (Fig. 5A, B). In conjunction with the effects of 2-APB, BAPTA and removal of extracellular calcium this indicates that DHA reduced the ATP-induced calcium influx via inhibition of store-operated calcium channels in HUVEC. In agreement with the present set of experiments, data from Ye et al. [9] indicate that DHA reduced the calcium influx induced by oxidative stress through displacement of TRPC1-a core component of the store-operated calcium channel protein from lipid rafts in the human endothelial cell line EAhy926 [24].
Beside other factors, nitric oxide may affect the function of store-operated calcium channels. Nitric oxide production in endothelial cells is regulated via eNOS expression and regulation of eNOS activity via calcium/calmodulin or eNOS phosphorylation [25]. In human endothelial cells eNOS activity is known to be regulated by phosphorylation at serine, threonine and tyrosine sites within the enzyme. The resulting enzyme activity depends on the phosphorylation at specific sites such as ser1177, ser615 residues for activation, and ser114 or thr495 residues for inhibition [26]. In the context of this study two phosphorylation residues of eNOS were studied, namely thr495 and ser1177. According to the generally accepted view, ATP by binding to its cell surface receptors transiently induces a rise of cell calcium influx. A more prolonged activation of eNOS activity may result from phosphorylation at ser1177 or dephosphorylation thr495 [272829]. In agreement with previous results, the current experiments show that ATP stimulation did not change total eNOS expression (Fig. 2A). Furthermore, ATP stimulation clearly increased eNOS phosphorylation at ser1177 residue (Fig. 2C), while eNOS phosphorylation at thr495 was unchanged (Fig. 2D). Long-chain polyunsaturated fatty acids including DHA enhance nitric oxide production and eNOS phosphorylation by Akt, ERK or cGMP-dependent manners [30313233]. Stebbins et al. [30] showed that chronic treatment of human coronary artery endothelial cells for 7 days with DHA in low-dose enhanced both eNOS phosphorylation at ser1177 and eNOS activity without changing total eNOS expression and NO production by Akt and cGMP-dependent manner. High dose of DHA could lead degradation of eNOS. This could explain for chronic treatment with low dose DHA to get beneficial effects on cardiovascular disease. In our study, I performed the experiment with ODQ-1 to inhibit the cGMP production, however, it did not show any effect on DHA-induced calcium transient. So we proposed that DHA reduced ATP-induced calcium transient by c-GMP independent manner. In line with our study, Chen et al. [31] explored that DHA increase NO production and Phosphorylated eNOS in cardiac fibroblasts without elevated cGMP level. Chao et al. [32] and Yamagata [33] proposed DHA inhibits VEGF-induced cell migration via eNOS activation through ERK pathway in HUVEC. Recently, Nuno et al. [34] suggested dietary fats (including DHA) could modulate lipid raft composition that could lead to affect eNOS localization and phosphorylation. In this study, we only tested for cGMP pathway due to calcium transient aspect. In addition, Li et al. [35] showed that HUVEC treated with DHA (25–50 µM) for 24 h doubled their eNOS activity. These authors suggested that the effect of DHA is mediated by a change of lipid raft composition resulting in the displacement of caveolin-1 and eNOS from caveolae. In another experimental study on the endothelial cell line EAhy926 reported that DHA at various concentrations (20–80 µM, 24 h) elevated eNOS phosphorylation at ser1177 [36]. Thus, there is ample evidence that in human endothelial cells low concentrations of DHA (50 nM–25 µM) activate eNOS activity. However, there is also evidence of a missing effect at higher concentrations of DHA (> 25 µM) [37]. The latter result might be a consequence of ROS generation via NADPH oxidase activation induced by high DHA concentrations in EAhy926 endothelial cells [38]. The current study using a DHA concentration of 12 µM (48 h) is in line with previous studies [39]. DHA increased ser1177 eNOS phosphorylation (Fig. 2E, F) and in addition slightly decreased thr495 eNOS phosphorylation (Fig. 2E, F). The effect of ATP induced eNOS phosphorylation appeared independent of the presence or absence of DHA. Thus, DHA treatment may enhance baseline eNOS activity without affecting short term regulation of eNOS activity evoked by stimulation of endothelial cells by ATP. Both phosphorylation changes at ser1177 and thr495 support an elevation of eNOS activity as shown in previous studies [3035]. It must remain open at present, whether modulation of these phosphorylation sites is associated with the beneficial effects (endothelial relaxation, coronary artery disease etc.) attributed to eNOS activation after DHA treatment [34]. BAPTA was used to chelate intracellular calcium that markedly inhibited ser1177 eNOS phosphorylation and calcium influx activated bradykinin-induced ser1177 eNOS phosphorylation [29]. In the current study, 2-APB, a blocker of store-operated calcium channels was used to inhibit calcium influx that could markedly reduce ATP-induced phosphorylation at thr495 (Fig. 4C–F). In addition, the non-specific P2R blocker PPADS, which reduced the ATP-induced intracellular calcium increase (including both calcium influx and calcium release from stores) (Fig. 6A, B), tended to reduce ser1177 eNOS phosphorylation (Fig. 6C), and significantly elevated thr495 eNOS phosphorylation induced by ATP (Fig. 6D). From these data it is concluded that ATP-induced phosphorylation of ser1177 eNOS most likely did not depend on calcium influx via store-operated calcium channels. Rather an intracellular calcium increase related to calcium liberation from intracellular stores may be functionally important. With respect to ser1177 phosphorylation the current study documents for the first time that dephosphorylation of thr495 eNOS is induced by ATP, which depended on P2R activation (Fig. 6C). Furthermore, in human endothelial cells the phosphorylation state at thr495 greatly depended on the inhibition of store-operated calcium influx channels.
In summary, the current study indicates that DHA, likely by incorporation into the cell membrane, exerts an important modulatory function on store-operated calcium channels. Hence, in the presence of DHA the calcium transient in response to ATP stimulation is blunted. However, DHA also enhanced ser1177 phosphorylation of eNOS, which suggests enzyme activation. Thus, it appears that eNOS activity in presence of DHA is rather augmented via enzyme phosphorylation than by calcium stimulation. In conjunction with a previous study in which we reported enhanced ATP and ADP release from endothelial cells by DHA treatment [12], multiple effects of this omega-3-fatty acid may interact stimulating endothelial cell ATP release, which via autocrine P2Y receptor activation stimulates membrane calcium influx and simultaneously changes eNOS phosphorylation in a manner predictive of enhancing nitric oxide production. Here, a separate effect of DHA also resulting in ser1177 phosphorylation may augment ATP dependent stimulation. The elevation of eNOS activity might also be expected to act via nitric oxide as a negative feedback on calcium influx via store-operated calcium channels. From current evidences, we concluded that DHA elevated eNOS phosphorylation probably via inhibition of store-operated calcium channels proved by reduction of ATP-induced calcium transient in human endothelial cells. These results could explain for beneficial effects of DHA on vascular bed in physiological and pathophysiological conditions of cardiovascular diseases as well as pulmonary diseases like asthma.
Go to :
 XML Download
XML Download