

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Tauopathies, a class of neurodegenerative diseases that includes Alzheimer's disease (AD), are characterized by the deposition of neurofibrillary tangles composed of hyperphosphorylated tau protein in the human brain.1 However, the underlying mechanism of tauopathy remains unclear, and deciphering the process is crucial for developing effective biomarkers and identifying potential therapeutic targets of AD. Abnormal alterations of histone acetylation and methylation have been documented in patients with AD and in a transgenic animal model of AD.2 In early development, several epigenetic alterations may contribute to AD, whereas certain epigenetic shifts may occur downstream of AD pathology.2 In particular, histone acetylation is indicative of late onset cognitive loss, and has been used to develop biomarkers of AD.3
Tau phosphorylation is an important pathogenic event in AD. Targeted expression of human tau in the central nervous system (CNS) of Drosophila recapitulates several characteristics in human AD, such as neuronal loss in the form of prominent vacuoles, compromised life span, and locomotor and cognitive impairments.4 Therefore, to provide a useful AD model, transgenic Drosophila were generated using an FTDP-17 mutant form of human tau (tauR406W) that was subcloned downstream of the yeast upstream activator sequence (UAS),5 tissue-specific expression of the transgene was achieved by regulating the availability of Gal4 transcription factor with different Gal4 drivers.6
Analyses of tissues from AD patients and a transgenic mouse model of AD consistently show decreased histone acetylation.7 Accordingly, histone deacetylase (HDAC) inhibitors have been proposed as potential procognitive agents for the treatment of AD.891011 Indeed, Tip60 histone acetyltransferase (HAT) in human tau overexpressing Drosophila has been proposed to play a neuroprotective role in impaired cognition during early development of AD.121314 Reciprocally, Drosophila genetic screens have shown that HDAC6 null mutation and pharmacological inhibition can rescue tau-induced microtubule defects in muscles and neurons.15 Furthermore, histone methylation has a marked influence on synaptic plasticity and cognition.81016
Recently, Mastroeni and colleagues reported increased histone H3K4 trimethylation in the cytoplasm and significant colocalization with hyperphosphorylated tau tangles, but decreased in the nuclei of AD brains compared to non-demented controls.17 Moreover, the total levels of histone H3K9 dimethylation and HP1α were decreased and heterochromatin relaxed, resulting in aberrant neuronal gene expression in Drosophila, mouse, and human tauopathies.5 However, no studies have investigated the involvement of histone demethylases in tauopathies.
In this study, bioinformatics analyses revealed significantly higher expression of several Jumonji domain-containing histone demethylase (JHDM) genes18192021 in postmortem brain tissue from patients with AD than from non-demented controls. In addition, our results suggest that the downregulation of kdm4a (JHDM3A) expression may be a potential therapeutic target in AD, providing new insight into the role of histone demethylases in tauopathies.
METHODS
Bioinformatics analyses
Expression levels of histone demethylases in postmortem brain tissue from patients with AD were evaluated using the publically available Gene Expression Omnibus (www.ncbi.nlm.nih.gov/geo, GSE33000) (Table 1). The dataset included patients with AD (n = 310) and non-demented controls (n = 157). JHDM1A (10025907153_at probe), JHDM1B (10025912776_at probe), JHDM2A (10023810558_at probe), JHDM2B (10025911102_at probe), JHDM3A (10025904057_at probe) and JHDM3B (10025924555_at probe) mRNA expression levels were compared between groups using unpaired, two-sided Student's t-tests.

Drosophila strains and genetics
Fruit flies were maintained on standard media, and genetic crosses were performed at 25°C unless specified otherwise. All genetic crosses were performed at least in triplicate, and percentages represent the means of the replicates. The following Drosophila strains were used: UAS-kdm2 RNAi (P{KK101783}VIE-260B) (Vienna Drosophila RNAi Center [VDRC], Vienna, Austria), UAS-kdm3 RNAi (y[1] v[1]; P{y[+t7.7] v[+t1.8] =TRiP.HMJ22328}attP40) (Bloomington Drosophila Stock Center, Indiana University, Bloomington, IN, USA), UAS-kdm4a RNAi lines (w1118; P{GD9133}v32652 hereafter referred to as UAS-kdm4a RNAi and w1118; P{GD9133}v32650/CyO hereafter referred to as UAS-kdm4a RNAi2) (VDRC), UAS-kdm4b RNAi (P{KK102089}VIE-260B) (VDRC), elav-Gal4 lines (P{w[+mW.hs]=GawB}elav[C155] and P{w[+mC]=GAL4-elav.L}2/CyO, #B8765) (Bloomington Drosophila Stock Center), UAS-kdm4a-HA1FLAG2 (w; P{w[+mC]=[UAS-kdm4a-HA1FLAG2]} hereafter referred to as UAS-kdm4a+) (Jerry L. Workman, Stowers Institute for Medical Research, Kansas, MO, USA),22 BL2 chromatin reporter (y1·w*/Dp(3;Y)BL2, P{HS-lacZ.scs}65E) (Bloomington Drosophila Stock Center), UAS-tauR406W (Mel B. Feany, Harvard Medical School, Boston, MA, USA),5 GMR-Gal4 (Deborah A. Hursh, CBER/FDA, Silver Springs, MD, USA).23 The wild-type strain was w1118 (Seungbok Lee, Seoul National University, Seoul, Korea).24
Locomotor behavioral assay
Flies were collected in vials on the day of eclosion and then transferred without anesthesia to fresh vials every other day. The locomotor assay was performed in the afternoon on day 10. Ten flies were placed in each plastic vial marked with 15 cm height and gently tapped to the bottom. The percentage of flies that climbed to the height of 15 cm in 10 seconds at each consecutive 5-day interval was calculated for each genotype unless specified otherwise. The assay was performed for 20 days after eclosion.
Histochemical detection
For histochemical detection of β-galactosidase activity, larvae were heat-shocked for 45 minutes at 37°C, followed by recovery for 1 hour at room temperature. Larval brain lobes were dissected in phosphate-buffered saline (PBS), fixed in 3.7% formaldehyde in PBS for 5 minutes, washed in PBS, transferred to X-gal solution,25 and incubated overnight at 37°C. Larval brain lobes were mounted in 80% glycerol and examined using an Olympus DP71 microscope.
Statistical analyses
All data were analyzed using Microsoft Excel 2007 and expressed as the means and standard deviations. Continuous variables were analyzed using Student's t-tests when data were normally distributed. All statistical tests were two-sided. P values < 0.05 were considered to indicate statistical significance.
RESULTS
Increased expression of several JHDM genes in postmortem brain tissue from patients with AD
To investigate the potential relationship between alterations in the expression of JHDM genes and AD pathology in vivo, we used bioinformatics analyses to examine differences in the expression levels of JHDM1A/1B, JHDM2A/2B, and JHDM3A/3B, which have Drosophila homologs (Tables 1 and 2). The expression levels of JHDM1A, JHDM2A/2B, and JHDM3A/3B were significantly higher in postmortem brain tissue from AD patients than from non-demented controls (average log2 value, 0.0221 ± 0.00337 vs. −0.0333 ± 0.00451; 0.0064 ± 0.00516 vs. −0.0794 ± 0.00543; 0.00837 ± 0.00276 vs. −0.0179 ± 0.00342; 0.0471 ± 0.00251 vs. 0.0256 ± 0.00326; 0.1403 ± 0.0155 vs. −0.0220 ± 0.0171, respectively; P < 0.001), whereas the JHDM1B mRNA levels were significantly downregulated in brain tissues from AD patients (average log2 value, −0.1300 ± 0.00603 vs. −0.0161 ± 0.00826; P < 0.001) (Fig. 1).
Table 2
List of genes for the study

![]()
 | Fig. 1Increased expression of JHDM genes in postmortem brain tissue from patients with AD. Scatter diagrams representing mRNA expression levels of JHDM genes in postmortem brain tissue from patients with AD. Dot plots showing JHDM1A/1B, JHDM2A/2B, and JHDM3A/3B mRNA levels in non-demented healthy controls and AD patients.
JHDM = Jumonji domain-containing histone demethylase, AD = Alzheimer's disease.
*P < 0.001 between the indicated groups.
|
Knockdown of JHDM genes ameliorates the morphological deficit induced by overexpressing tauR406W in Drosophila eyes
To examine the effect of JHDM gene knockdown in an AD background, tauR406W-expressing flies were crossed with UAS-RNAi strains expressing RNAi specific for each of the JHDM fly homologs (kdm2, JHDM1; kdm3, JHDM2; kdm4a, JHDM3A; and kdm4b, JHDM3B) (Table 2) in Drosophila eye using the GMR-Gal4 driver. Neurodegeneration can be easily monitored when tauR406W is overexpressed in the fly eyes, resulting in eye morphological defects such as reduced size, loss of bristles, disordered ommatidia and roughened eye surfaces.2627 The observed eye defects can be categorized into four classes according to their severity: class 1, normal; class 2, roughness in the eyes with size reduction and some ommatidial disruption; class 3, collapsed eye tissue with size reduction; and class 4, black spots of apoptotic tissue in the collapsed eye (Fig. 2A). Approximately 74% of flies overexpressing tauR406W have class 3 + 4 eyes. In contrast, knockdown of kdm2, kdm3, kdm4a or kdm4b genes in tauR406W-overexpressing flies ameliorated the tauR406W-engendered eye defects, resulting in only 8%, 37%, 40%, or 21% class 3 + 4 eyes, respectively (Fig. 2B). No eye defects were detected in flies overexpressing each of the UAS-RNAi strains of kdm alone (data not shown). Therefore, knockdown of the kdm genes ameliorated the tauR406W-induced eye phenotype, resulting in less severe phenotypes.
 | Fig. 2Knockdown of any of the JHDM genes ameliorates the morphological defects induced by overexpressing tauR406W in Drosophila eyes. (A) Bright field images of 10-day-old adult Drosophila eyes of control (w1118) and tauR406W transgenic flies heterozygous for knockdown of any of the kdm genes using the GMR-Gal4. Images were taken using an Olympus SZ61 stereo zoom binocular microscope equipped with an eXcope XCAM1080 digital camera. (B) kdm knockdown flies that overexpressed tauR406W showed a shift toward less severity in the distribution of phenotype categories in a population of flies (n = 30–73).
JHDM = Jumonji domain-containing histone demethylase.
|
Targeted downregulation of kdm4a ameliorates tauR406W-induced locomotion defect
To evaluate the neuronal effect of kdm gene knockdown in flies overexpressing tauR406W in their CNS via pan-neuronal elav-Gal4, we performed the climbing assay, a behavioral readout used for monitoring neurotoxic deficits in Drosophila.2829303132 Whereas w1118 control flies retained 90% of their climbing ability on day 10, only 45%–50% of tauR406W-expressing flies climbed up. Inconsistent with the eye analysis results, knockdown of either kdm2 or kdm3 in flies overexpressing tauR406W had no significant effect on the climbing ability of flies overexpressing tauR406W alone (Fig. 3A and B). Interestingly, flies that overexpressed kdm2 RNAi alone exhibited a locomotion defect, showing 60% climbing ability compared to 90% climbing ability in control flies. This finding indicates that kdm2 itself may play a role in neuronal function. However, knockdown of kdm4a in tauR406W-overexpressing flies caused a mild but significant amelioration of the locomotion defect, with flies showing 80% climbing ability compared to the 50%–60% climbing ability of flies overexpressing tauR406W alone (Fig. 3C). Importantly, overexpression of kdm4a RNAi alone had no significant effect on locomotion, indicating that the neuroprotective effect may be specific to the context of tauopathy. In contrast, knockdown of kdm4b in flies that overexpressed tauR406W exacerbated the locomotion defect, and they showed 30% climbing ability compared to 50% in flies overexpressing tauR406W alone. Moreover, flies expressing kdm4b RNAi alone exhibited the locomotion defect, showing 60% climbing ability compared to 90% in control flies, indicating that kdm4b itself may be involved in neuronal function (Fig. 3D). Therefore, tauR406W overexpression may aggravate the neuronal defect in kdm4b-deficient flies during development.
 | Fig. 3Targeted downregulation of kdm4a ameliorates tauR406W-induced locomotion defect. (A–E) Locomotor activity in control and tauR406W transgenic flies heterozygous for knockdown of any of the kdm genes using pan-neuronal elav-Gal4 (#8765) was measured on day 10 post-eclosion (10 flies and 10 repeats per group, respectively; 15 cm/10 sec). (F) Locomotor activity in control and tauR406W transgenic flies heterozygous for overexpression of wild-type kdm4a transgene using pan-neuronal elav-Gal4 (#c155) was measured on day 7 post-eclosion (10 flies and 10 repeats per group, respectively; 8 cm/20 sec). (G) A shift in the distribution of eye phenotype categories in populations of flies with kdm4a knockdown or overexpression in tauR406W-overexpressing flies using GMR-Gal4 (n = 78–89).
*P < 0.05 and n.s. denotes P > 0.05 between the indicated groups.
|
To ascertain the neuronal protective effect of kdm4a knockdown in tauR406W-overexpressing flies, we performed the climbing assay with different kdm4a RNAi strain or wild-type kdm4a transgenic flies crossed to tauR406W-overexpressing flies. Crossing the kdm4a RNAi2 line with flies overexpressing tauR406W in their CNS ameliorated the locomotion defect, as they showed 70% climbing ability compared to 50% in flies that overexpressed tauR406W alone (Fig. 3E), whereas exogenously overexpressing wild-type kdm4a in tauR406W-overexpressing flies exacerbated the locomotion defect, as they showed 10% climbing ability compared to 40% in flies overexpressing tauR406W alone (Fig. 3F). Accordingly, in the eye, kdm4a knockdown with the RNAi2 strain in tauR406W-overexpressing flies resulted in 58.5% class 3 + 4 eyes compared to 87% class 3 + 4 eyes in flies overexpressing tauR406W alone, whereas overexpressing kdm4a in tauR406W-overexpressing flies showed no significant difference in eye morphology compared to those overexpressing tauR406W alone (Fig. 3G). Taken together, kdm4a knockdown ameliorated tauR406W-engendered deficits in both neurons and eyes.
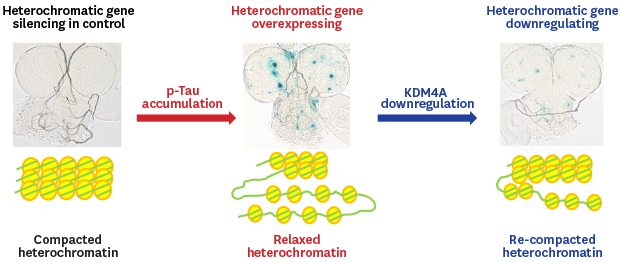
Targeted downregulation of kdm4a reduces heterochromatic loss induced by tauR406W overexpression
Given that tau promotes neuronal death through heterochromatin loss, global chromatin relaxation, and aberrant transcriptional activation in Drosophila, mouse, and human tauopathies,5 we investigated whether knockdown of kdm genes alters heterochromatin loss in flies overexpressing tauR406W in their CNS via pan-neuronal elav-Gal4. We measured reporter expression in larval brain lobes using a transgenic fly strain (BL2 reporter) that has a LacZ reporter gene embedded in and silenced by heterochromatin.533 After 45 min heat shock pulse at 37°C, this LacZ reporter was silenced in controls (Fig. 4A), but abnormally expressed in tauR406W-overexpressing fly brain due to heterochromatin loosening (Fig. 4B). kdm2 knockdown enhanced the expression of the heterochromatin-embedded reporter compared to overexpressed tauR406W alone (Fig. 4C and F). Expectedly, RNAi-mediated reduction of kdm4a decreased the number and intensity of the expression foci of the heterochromatin-embedded reporter in tauR406W-overexpressing flies (Fig. 4D and F), whereas kdm4b knockdown had no significant alteration on the expression of the heterochromatin-embedded reporter in flies overexpressing tauR406W alone (Fig. 4E). Therefore, neuronal-specific downregulation of kdm4a may suppress tauR406W-engendered locomotion impairment by restoring heterochromatin.
 | Fig. 4Targeted downregulation of kdm4a reduces heterochromatin loss induced by tauR406Woverexpression. (A–E) The lacZ expression (BL2 reporter) embedded in and silenced by heterochromatin displayed in larval brain lobes of each of the indicated genotypes driven by pan-neuronal elav-Gal4 (#8765) under heat shock pulse (45 min at 37°C), using X-gal staining. (F) Quantification of BL2 chromatin reporter expression of indicated genotypes (n = 5–10).
*P < 0.05 and n.s. denotes P > 0.05 between the indicated groups. Scale bar = 100 μM.
|
DISCUSSION
We used bioinformatics analyses to detect alterations in JHDM gene expression in AD pathology. The expression levels of JHDM1A, JHDM2A/2B, and JHDM3A/3B were significantly higher in postmortem brain tissue from AD patients than from non-demented controls. In Drosophila, CNS-specific knockdown of kdm4a (JHDM3A) ameliorated tauR406W-induced locomotion defects, in agreement with eye analysis results.
Recently, Frost et al.5 reported that oxidative stress and subsequent DNA damage can substantially alter chromatin structure, inducing heterochromatin loss and aberrant transcriptional activity, causing neurodegeneration in tauopathies. The authors reported that tauR406W-overexpressing adult fly brain had decreased total levels of H3K9 dimethylation and heterochromatin protein 1α (HP1α), which are normally enriched in heterochromatin. In addition, loss of function mutations in HP1α or Su(var)3-9, which encodes a histone methyltransferase responsible for H3K9 dimethylation, exacerbated tauR406W-engendered locomotion deficits. Given that KDM4A also directly interacts with HP1α and is essential for heterochromatin organization and function via both enzymatic and structural mechanisms in Drosophila,223435 it is plausible that KDM4A may be associated with heterochromatin alteration in tauopathies. Intriguingly, kdm4a knockdown with either of two RNAi strains ameliorated tauR406W-induced deficits in the eye and in neurons. Conversely, wild-type kdm4a overexpression in flies overexpressing tauR406W in the CNS exacerbated tauR406W-induced locomotion defects. Importantly, overexpression of kdm4a RNAi alone had no significant effect on locomotion in flies, indicating that the neuroprotective effect was specific for the AD pathological context. Consistent with these results, Lorbeck et al.36 reported that flies containing the P-element suppressor mutant of kdm4a (kdm4aP-supp) showed no significant loss in climbing ability. According to the BL2 transgenic reporter assay, RNAi-mediated reduction of kdm4a in the CNS reduced heterochromatin relaxation, thus ameliorating the tauR406W-engendered locomotion defect. Further studies are needed to elucidate the underlying mechanism of KDM4A involvement in the heterochromatic organization and transcriptional regulation of tauopathies.
kdm4b knockdown in the CNS exacerbated the tauR406W-induced locomotion defect. Interestingly, flies that overexpressed kdm4b RNAi alone exhibited the locomotion defect, showing 60% climbing ability compared to 90% in control flies. This finding indicates that KDM4B may play a role in neuronal function in Drosophila. Consistent with this result, neuronal-specific kdm4b-deficient mice exhibit hyperactive behavior, sustained hyperactivity in a novel environment, deficits in working memory, and spontaneous epileptic-like seizures, indicating impaired neurodevelopment.37 This finding suggests that KDM4B plays a crucial role in the formation of functional neuronal networks. Therefore, neuronal defects in kdm4b-deficient flies during development may exacerbate tauR406W-engendered locomotion impairment. However, the underlying mechanism does not appear to involve altered heterochromatin structure, as kdm4b knockdown had no significant effect on heterochromatin relaxation induced by tauR406W overexpression.
Inconsistent with the eye analysis results, knockdown of either kdm2 or kdm3 in tauR406W-overexpressing flies had no significant effect on climbing ability in flies that overexpressed tauR406W alone. However kdm2 knockdown tended to slightly exacerbate the tauR406W-induced locomotion defect, particularly in young flies (2–3 days after eclosion), but not significantly in older flies (data not shown). Furthermore, flies expressing kdm2 RNAi alone exhibited the locomotion defect, showing 60% climbing ability compared to 90% in control flies, indicating that KDM2 may play a role in neuronal function. In agreement with this result, kdm2 null mutant adult flies displayed defects in circadian locomotor behavior.38 Thus, a neuronal defect in kdm2-deficient flies during development may exacerbate the tauR406W-engendered locomotion impairment in young flies. Furthermore, kdm2 knockdown in the larval brain lobe can enhance the number and intensity of expression foci of the heterochromatin-embedded reporter in tauR406W-overexpressing flies. These results indicate that neuronal-specific downregulation of kdm2 may exacerbate tauR406W-induced locomotion impairment by enhancing heterochromatin loss during early development; however, the underlying mechanism remains to be elucidated.
Using bioinformatics analyses, we revealed significantly increased expression of JHDM1A, JHDM2A/2B, and JHDM3A/3B in postmortem brain tissue from patients with AD compared to non-demented controls, whereas JHDM1B mRNA levels were downregulated in the brains of AD patients. Using tauR406W-induced transgenic flies as an AD model, we showed that kdm4a knockdown in the CNS ameliorated tauR406W-induced locomotion defects by restoring heterochromatin. These results suggest that the downregulation of kdm4a expression could be a potential therapeutic target in AD.
 XML Download
XML Download