

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Current definition of EoE
Eosinophilic Esophagitis (EoE) is a food allergy-associated inflammatory disease affecting both children and adults [12]. EoE is characterized by persistent esophageal inflammation due to an immune-allergic pathogenesis with clinical manifestations relating to esophageal dysfunction. Eosinophil accumulation and infiltration of esophageal mucosa results in histological changes leading to dysphasia, stricture and food impaction [12].
Diagnoses of EoE have grown rapidly in recent 5 years. In Western countries, the prevalence of EoE has increased dramatically from 10 to 50 for every 100,000 general population [3]. Meanwhile, albeit published reports of EoE from Asian countries are limited, systematic literature review of EoE in Asian countries by Kinoshita et al. [4] reveals comparable EoE prevalence amongst Western and Asian patients. As a matter of fact, EoE is now becoming a serious disease not only in Western countries but also in Asia [4]. In industrialized countries, the incidence of EoE has increased dramatically in the past 30 years, resulting in a considerable public health and economic burden [2]. In many patients, the disease can be controlled by adherence to an elemental diet eliminating foods that trigger EoE [2]. However, this approach involves disruptive changes in lifestyle and eating habits [2].
Generally, the types of EoE have been characterized as food allergies. Around 70% of EoE patients have current or past food allergy history or positive skin pricks test particularly to a range of foods. The basis for this understanding commenced approximately 20 years ago, when Kelly et al. [5] reported that children with EoE, treated with a nonallergenic amino acid-based formula diet for 6 to 8 weeks, had a complete clinical resolution. However, reintroduction of the food proteins was associated with a clinical relapse [5]. Markowitz et al. [6] replicated these results in 2003 with a larger cohort. Recent clinical trials with practical elimination diet [7] and skin testing directed elimination diet [8] provided additional evidence of EoE as a food allergen driven condition. Previous study found that children with EoE were mainly sensitized to egg, dairy milk, soy, beef, wheat, and peanuts [9]. In a pediatric EoE cohort, sensitizations to food allergens have been identified in 75% of the study population [10], supporting the role of food allergy in EoE. Noteworthy is the observation of local immunoglobulin E (IgE) in esophageal mucosa of pediatric EoE patients [11]. In adult and pediatric patients with EoE, specific IgE to food and inhalant allergen epitopes have been recognized in 91% of the subpopulation [12], further supporting a role for food allergic mechanisms in EoE.
EoE also has been recognized as a connection between food allergy and other atopic diseases. EoE is strongly associated with atopic disease and frequently occurs together with asthma, eczema, allergic rhinitis, and anaphylactic food hypersensitivity, and a strong atopic family history [1013]. Allergy-prone children can begin with allergic sensitization, often with eczema in the first year of life, later developing allergic rhinitis and can progress to asthma [14]. This stepwise increment of conditions has been well described as the 'atopic march' [14]. In particular, food allergic children with multiple allergies, for example, asthma, eczema, or rhinitis at a young age are more likely to develop EoE [13]. Recent study has discovered that up to half of food allergy patients who have other atopic illnesses may progress to EoE later in life [10]. Interestingly, while these allergic diseases can follow the "atopic march," EoE shows a wide incident age range, and thus appears not to follow this established concept. Nonetheless, the strong clinical connection between EoE and other well characterized allergic diseases appears to suggest shared pathogenetic mechanisms with mutual impact on severity. As the result, EoE is therefore increasingly seen as a syndrome rather than a single disease [15]. There is also extensive clinical overlap of EoE with asthma, the immunological basis of which has been reviewed [16].
As with many chronic inflammatory diseases, clinicians now realize that the division of EoE into IgE-mediated and non-IgE-mediated food allergy has been an oversimplification. The pathogenesis differs in terms of genetic susceptibility, environmental risk factors, age of onset, clinical presentation, prognosis and response to standard and new therapies [17]. Therefore, in this review, we will focus on the underlying immunological basis of the various EoE pathogenetic mechanisms, specifically about how food allergens induced EoE, integrating results from human studies focusing on specific pathways with results from animal studies, where much molecular data are available.
Different types of food allergy-induced EoE
A number of clinical studies report that food allergy may be the cause of EoE pathogenesis [678910]. Most patients with EoE respond to an elemental diet with resolution of symptoms and normalization of biopsies [12]; in many cases, reintroduction of foods routinely causes relapse of esophageal eosinophilia [9]. The most widely known food allergies identified are linked to eggs, soy, chicken, beef, wheat, rice, barley, peas, corn, and peanuts [9]. It still remains unclear why these foods are more prone to trigger EoE than others, however there is an evidence that specific foods possess essential immunological properties that can induce intrinsic immune responses. For example, milk sphingomyelin can activate invariant natural killer T (iNKT) cells promoting Th2-response [18], and peanut allergen Ara h1 can bind to CD209 on dendritic cells (DCs) [19]. In addition, clinical reports demonstrate that peanuts or other nuts are attributed to 80% of fatal food allergy reactions [20], and the combined skin prick and patch-test-positive dataset of the Cincinnati Center for Eosinophilic Disorders specifies that patients with EoE most generally have peanut, corn, and sensitivities (unpublished data, stated in [21]). In humans, elimination and reintroduction diets imply the causal nature of foods in EoE [9], as does intragastric ovalbumin promoting EoE in mice [22]. This diet involves eliminating the 6 most common foods that have been recognized in allergic diseases, which are egg, milk, soy, wheat, nuts, and seafoods [9]. Elimination diet is initially suggested by a gastroenterologist as a result of referral to allergists to perform a food allergy evaluation for EoE. Eighty-eight percent resolution is observed in children with EoE after eliminating these foods from their diets [923]. Significant changes are required in daily diet and lifestyle and supplementation with an amino acid-based formula, which has been shown to produce clinical improvement in EoE patients [5]. The restricted number of foods that can promote EoE suggests there are certain essential immunological properties of these food proteins that can promote allergenicity [24], although this remains controversial. Taken together, the essential properties of certain food proteins, barrier dysfunction, and immune dysregulation susceptibility affected by early life events disposing to immune dysregulation, all contribute to sensitization.
The immunology of food allergy-induced EoE
Innate immune cells are important mediators of EoE pathogenesis. These cells include various type of T cells particularly Th2 cells, NKT cells, innate lymphoid cell (ILC) cells, and other inflammatory cells. They are individually discussed below including how they work together to promote food allergy-induced pathogenesis of EoE.
Eosinophilic esophagitis as a Th2 disorder
Within the field of chronic allergic inflammation, eosinophilic esophagitis is viewed as the trademark Th2 disorder of esophagus [25]. This is supported by the early findings that inflammation in esophageal biopsies of EoE patients is frequently eosinophilic in nature [568]. Initial studies demonstrated elevated numbers of CD4 positive cells producing interleukin (IL)-4, IL-5, and IL-13 in peripheral blood mononuclear cells (PBMC) [26] and esophageal mucosa biopsies of EoE patients [1227] correlating with the amount of esophageal eosinophilia [27]. Subsequent unbiased clustering algorithms have demonstrated that the various EoE phenotypes fall into Th2-prevalent immune response as the cause of the elevation of the cytokines IL-4, IL-5, and IL-13 and eosinophils in blood and tissue [122527]. Current studies indicate that there are food-specific Th2 cells in the peripheral blood of adult and pediatric patients with EoE [28]. Consistent with the IL-5 dependent and IgE-independent disease mechanisms for eosinophilic gastrointestinal syndromes [29], EoE subjects have food-specific Th2 cells that produce IL-5 as opposed to subjects with food anaphylaxis in whom food-specific Th2 cells generate IL-4, an interleukin which is vital for IgE class switching [28]. When cultured, IL-5 positive cells need longer time and chromatin remodeling to develop [30], in line with the chronic nature of the EoE disease process. If demonstrated in the periphery of EoE subjects, these cells could offer understanding into disease pathogenesis and offer a potential alternate allergic sensitization marker. In addition, there may be better alternate biomarkers of the Th2 high endotype of EoE, such as increased mRNA levels of eotaxin-3, IL-5, IL-5 receptor α-chain, and IL-13 that are associated with increased numbers of esophageal mucosa tissue eosinophilia [122627]. The case for CD4 positive Th2 cells as controllers of the EoE disease was therefore confirmed (Fig. 1).
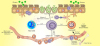 | Fig. 1EoE exhibits features of a T-helper type 2 (Th2)-predominant inflammation. Antigen is captured by antigen presenting cells, which induces a Th2-response in eosinophilic esophagitis. Interleukin (IL)-5 stimulates the proliferation and differentiation of eosinophils in the bone marrow, retains their survival and enables their migration into the blood. IL-13 facilitates eotaxin-3 generation from the epithelial cells, which trafficking eosinophils to the esophagus. Eosinophils generate Th2-cytokines circulating the inflammatory cycle and, thus, release cytotoxic granules initiating tissue injury. They induce dendritic cells activation via eosinophil derived neurotoxin to trigger Th2-cells response. Eosinophils via major basic protein prime mast cells activation. Mast cells can also be actuated by antigens cross-linking their surface IgE. Activated mast cells release IL-13, IL-5 and other pro-inflammatory mediators. Together, mast cells and eosinophils cause tissue remodeling and inflammation. ICAM, intercellular adhesion molecule; VCAM, vascular cell adhesion molecule.
|
The mice that lack the key Th2 cytokines (IL-4, IL-5, or IL-13) all have considerable reduction in esophageal eosinophilic features in the EoE model [31]. By analyzing mice overexpressing IL-5, either by hereditary manipulation or by IL-5 administration, researchers established the specific role of IL-5 in recruiting circulating eosinophils to the esophagus [3132]. Noti et al. [33] demonstrated mRNA levels of IL-4, IL-5, IL-13 in esophageal biopsies of EoE mice supporting that the EoE model depends strongly on Th2.
In addition, in the peanut oral sensitization induced EoE disease mouse model, characterized by esophageal inflammation, there is increased generation of peanut-specific IgE, and elevated peanut protein stimulated secretion of IL-4, IL-5, and IL-13 in splenocyte cultures [21]. Moreover, CD8-deficient mice are prone to experimental EoE whereas CD4-deficient mice are partly protected from esophageal inflammation [31].
There is randomized controlled trial using an antibody to the α-chain of the receptor for IL-4 (dupilumab; Regeneron Pharmaceuticals Inc., Eastview, NY, USA), which effectively inhibits downstream signaling via the receptors for IL-4 and IL-13 to study the effect of this treatment in adults with active EoE (https://clinicaltrials.gov/ct2/show/NCT02379052). Inhibitors of IL-13, such as the anti-IL-13 antibodies lebrikizumab or QAX576, may be a potential therapeutic choice [34]. In a phase 2 trial, blocking IL-13 with lebrikizumab (Genentech Inc., South San Francisco, CA, USA) show promising results, in reducing intraepithelial esophageal eosinophil counts and improving clinical symptoms in adults with EoE [34]. However, notwithstanding the results of an early safety trial of mepolizumab administration demonstrating a symptomatic improvement and a dramatic reduction in peripheral and esophageal eosinophilia in 4 patients with EoE [35], subsequent placebo-controlled and/or double-blind studies have failed to exhibit clinical effectiveness of mepolizumab or reslizumab in EoE pediatric patients, in spite of decrease in blood and esophageal eosinophilia [3536]. Regardless of whether the failure of clinical response reflects inadequate depletion of tissue eosinophils, irreversible structural changes, or involvement of other cell lineages or variables unaffected by anti-IL-5 therapy is uncertain. A randomized, double-blind, placebo-controlled, trial of an antibody to the receptor for IL-5 (benralizumab; MedImmune, a subsidiary of AstraZeneca, Gaithersburg, MD, USA) that effects depletion of eosinophils for months after a single injection is still ongoing (http://www.smartanalyst.com/SMART_Immunology_Newsletter.pdf).
ILCs in EoE
In human studies, IL-4 or IL-5 receptor blockade produces a positive clinical response in patients with esophageal eosinophilia irrespective of atopic status. In a knockout mouse model of EoE, esophageal eosinophilia is abrogated in RAG1 deficient (lacking B and T cells) or FOXN1 deficient (lacking T cells) mice, but not in IgH6 deficient (lacking B cells) mice [31]. These murine models support the view that Th2 cytokines and eosinophilia can be produced independent of the adaptive immune system.
ILCs were initially categorized as either non-T or non-B effector cells in various disease models of Th2 immunity [37]. Their nomenclature has been further delineated according to their cytokine production profile, into group 1 ILCs (interferon-γ producing which include traditional NKT and ILC1 cells), group 2 ILCs (Th2 cytokines producing, formerly known as ‘nuocytes’ or ‘natural helper cells,’ now named ‘ILC2 cells’), and group 3 ILCs (IL-17 and/or IL-22 producing, or are involved in the formation of lymphoid tissues, which include ILC3 cells and lymphoid tissue–inducer cells) [37]. ILC2 cells resemble Th2 cells in various ways as shown in Fig. 2. ILC2 cells lack antigen-specific receptors, but similar to Th2 cells, they respond to the epithelial derived cytokines IL-25, IL-33 and thymic stromal lymphopoietin (TSLP) [253337]. Study indicates that ILC2 cells development relies on IL-7 and IL-33, which is produced mainly from common lymphoid progenitors [37].
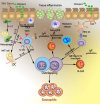 | Fig. 2Comparative roles of T-helper type 2 (Th2) cells and type 2 innate lymphoid cells (ILC2) cells in 2 forms of eosinophilic esophagitis (EoE). Eosinophilic inflammation and tissue remodeling are compelled by adaptive Th2 cells that are stimulated by dendritic cells (DCs) to generate interleukin (IL)-5, IL-13 and IL-4, the latter driving IgE and IgG synthesis. ILC2 reacts to epithelial cytokines thymic stromal lymphopoietin (TSLP), IL-33, and IL-25 and in thusly deliver Th2 cytokines. ILC2 IL-4 play a role in contributing to Th2 cell differentiation. Additional ILC2 incitement may take place from prostaglandin D2 and leukotriene D4 mediators produced by mast cells and macrophages, and inhibitory signals from lipoxin A4. Th2 and ILC2 IL-5 generation promotes eosinophil activation and survival, whereas Th2 and ILC2 IL-13 leads to tissue remodeling, and along with IL-9, induces mucus production. Together, the responses triggered by secretion of type 2 cytokines from both ILC2 and Th2 cells orchestrate EoE allergic inflammation and tissue remodeling. NKT, natural killer T; MHC, major histocompatibility complex; TCR, T-cell receptor.
|
Activation of ILC2 cells is further stimulated by TSLP, IL-25, and IL-33, produced mainly by epithelial cells, as a response to injury and stimulation via pattern-recognition receptors [3238]. Culture studies with lymphoid aggregate cells from EoE human donor esophageal tissue in the presence of TSLP or IL-33 cytokine induce ILC2 proliferation. Thus, ILC2 cells are detected in the esophageal mucosa patients with EoE and respond to cytokines recognized to stimulate ILC2 proliferation [3940]. Clinical studies also demonstrate elevated levels of IL-25, IL-33, TSLP in active EoE as compared to normal esophageal tissue [3840]. In addition, ILC2 cells producing Th2 cytokines (IL-13, IL-5, and IL-9) were initially described in the human esophageal EoE tissue, where they contribute to esophageal tissue eosinophilia and play a fundamental role in inflammation [40]. Although ILC2s lack surface markers for B, T, or NKT cells, they do express homologous chemo-attractant receptor molecules commonly expressed on Th2 lymphocytes (CRTH2) [39]. In line with the rapid increase in knowledge about ILC2 cells, including experimental models of Th2 allergic disease and clinical studies of ILC2 cells in pediatric EoE [38], the contributions of ILC2 cells relative to that of Th2 cells in EoE have emerged.
ILC2 cells may have an important role in a subset of EoE patients with lack of or partial response to steroids [41]. IL-5 and IL-13 secretion are not downregulated by steroids in ILC2 cells as they are in CD4 positive T cells. TSLP can trigger steroid resistance in ILC2 cells by up-regulating the pro-survival factor Bcl-xL and promoting phosphorylation of the transcription factor STAT5 [42]. Hence, if ILC2 cells are pathogenic in this subset of patients, this would help explain steroid resistance.
REGULATORY T (T REG) CELLS IN EoE
For many years, it was hypothesized EoE may develop because patients have a deficiency in normal or activated Treg cells [4344]. Tregs, characterized by FOXP3 expression, are essential for the development of tolerance. Results from pediatric and adult clinical studies are inconsistent with respect to Treg numbers in EoE with high Treg expression in pediatric cases, but a relative lack in adults [43] which may indicate a difference between pediatric and adult EoE disease. Furthermore different results between animal and human studies of Tregs may be due to differences in study design [15434445]. Thus, the exact role of Treg cells in human with EoE diseases remains controversial.
In EoE patients, the numbers and activity of Treg cells esophageal tissue is reduced compared to healthy subjects [43] although there is inconsistency as esophageal Treg cell numbers are elevated in children [4446] in some studies but diminished in adults in others [43]. Despite these discrepancies, all studies concur that Treg functional activity is reduced in EoE [43]. Interestingly, this reduction in Treg cell function in EoE patients appears to affect only the regulation of Th2 responses with the elevated numbers of CD4 positive effector T cells [46].
In the blood of EoE children, a subpopulation of Treg cells coexpressing the transcription factors Foxp3 and GATA-3 could suppress production of Th1 but not Th2 cytokines by PBMC [44]. This suggests that Treg cells in EoE patients might promote allergen-specific Th2 responses rather than suppress them. These Treg cells block TIGIT expression and generate fibrinogen-like protein 2, which spares Th2 responses while regulating Th1 and Th17 reactions [47] as shown in Fig 3. Regardless of whether this TIGIT positive population and the GATA-3 positive Foxp3 positive T cells found in EoE are linked or not remains unclear. Further studies are required to elucidate the roles of both Tregs and T cells in EoE.
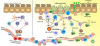 | Fig. 3Abnormal interactions between allergen and the host immune system in gastrointestinal tissue in eosinophilic esophagitis (EoE) pathogenesis. Antigen presenting cells present food allergens as major histocompatibility complex class II conjugates to T-cell receptors on naive T cells. T cells then differentiate into T-helper type 1 (Th1) or Th17 cells, which secrete different pro-inflammatory and anti-inflammatory cytokines leading to recruitment of humoral and cellular factors of innate immunity. In healthy tissues (left), immune homeostasis is maintained by interdependent control exerted by sufficient Treg cells. In mucosal response in EoE (right), dysfunctional regulation of Th1 or Th17 pathways triggers an unregulated inflammatory response and recruitment of innate immune cells. Cytokine levels elevation can promote oxidative tissue damage and facilitate proteolytic peptides and enzymes recruitments, eventually presenting as gastrointestinal disease. Furthermore, enactment Th2 pathway can prompt plasma cells identifying fungal cell wall antigens and delivering antiglycan antibodies production. IL, interleukin; NKT, natural killer T; ICAM, intercellular adhesion molecule; VCAM, vascular cell adhesion molecule.
|
A MIXED Th1 AND Th17 DISORDER IN EoE
Patients with late-onset and more intense allergic inflammation appear to have a mixed Th1 and Th17 cytokine milieu for supporting the development of IL-17 positive Th cells [4849]. The role of IL-17 and Th17 cells in EoE still remains unclear, with a recent study supporting the role of Th17 cells in the pathogenesis of EoE [50]. Comparable outcomes have been obtained for IL-23, another cytokine generated by Th17 cells [51]. Another study indicated that IL-17 elevation in PBMCs of active EoE patients. Meanwhile, there seems to be an age-related variation in the expression of IL-17 between children and adults with active disease [50]. In addition, there is also a complex interaction between Th17 cell–and tumor necrosis factor (TNF) or transforming growth factor beta (TGF-β) which promote differentiation of Th17 [49] as summarized in Fig. 3.
Both TGF-β and TNF-α are assumed to have a pivotal role in EoE pathogenesis. Polymorphisms in the agent for TGF-β1 are related to EoE predisposition, and TGF-β1 positive cells are overexpressed in the esophagus of EoE patients [5253]. Similarly, TNF-α is upregulated in EoE and is overexpressed by esophageal epithelial cells in active EoE patients [1542]. TGF-β1 is secreted by eosinophils and mast cells and promotes collagen production and tissue fibrosis by regulating Smad3 pathway signal transduction [54]. Consistent with this, esophageal fibrosis and angiogenesis are blocked in Smad3-deficient mice [22]. It is likely that the activated epithelial cells prime esophageal fibroblasts to secrete profibrogenic cytokines IL-1β and TNF-α, which drives epithelial-to-mesenchymal transition and esophageal fibrosis [52]. In one study, coculture of primary epithelial or muscle cells derived from patients with EoE increases cell line secretion of fibronectin and collagen I, which is inhibited through blocking TGF-β1 [53]. Coculture of eosinophils with cultured muscle cells also resulted in reduced contractility, an observation that may be the result of TGF-β1-induced phospholamban expression [53].
In view of these observations, it is postulated that blocking TGF-β1 can be a promising treatment for EoE therapy. One study found following treatment with TGF-β1 inhibitor, there was nearly a 50% reduction of contraction in smooth muscle cells derived from esophageal tissue of EoE patients [55]. Blocking TNF-α with infliximab (a chimeric IgG1 mAb directed against TNF-α) may also be effective in the treatment of EoE patients, although in a small study of 3 adult EoE patients, infliximab therapy did not show a significant benefit [56]. However, in view of the small numbers in this study, further studies of TNF-α blockade in EoE are needed.
Th9 CELLS IN EoE
IL-9 is formerly known as a Th2-specific cytokine promoted by IL-2, IL-4, and TGF-β [57]. Th9 cells have been distinguished as a particular Th cell subset, and another study has suggested that supplementary stimuli such as TSLP, signaling through Notch family receptors or OX40 receptor ligation are likely to support Th9 cells differentiation [58]. Studies of molecular pathways in IL-9 generation have observed that binding of the Smad2, Smad3, STAT5, IRF4 and PU.1 transcription factors to the IL9 agent are vital for Th9 polarization and IL-9 production [5759]. This process is influenced by the inhibitory SOCS proteins, and deficiency in CIS, a member of the SOCS family, causes increased Th2 differentiation and contributed to EoE pathogenesis [60].
In addition, IL-9 can also act as a potent mast cell growth factor that supports IL-4 regulated antibody generation by B cells and goblet cell metaplasia production [59]. IL-9 is highly expressed in the esophageal tissues of patients with EoE allowing this pleiotropic cytokine to facilitate mast cells activation and maturation [60]. Notably, IL-9 gene transcription is prominent in esophageal tissue of EoE patients [61]. Results showing that decrease in esophageal mast cell numbers occur predominantly in a subgroup of patients EoE who respond to the anti-IL-5 antibody treatment raises the issue of the cellular origin of IL-9 [62]. A double-immunofluorescence analysis study indicates that major basic protein (MBP) positive eosinophils and other undisclosed neighboring cells with tryptase positive mast cells in the esophagus secrete IL-9. This intriguing observation implies that the esophageal eosinophils are only one of the IL-9 generating immune cell types involved in the pathogenesis of EoE. On the other hand, despite that T cells were previously considered as the primary origin of IL-9, experiments with fate-mapping technique reporter genes in transgenic mice have shown that, following papain administration, ILC2 cells produce more IL-9 than do T cells [63].
IL-9 derived from ILC2 cells has been shown to be an autocrine enhancer of ILC2 cell functioning by promoting their survival [62]. ILC2 transiently secrete IL-9 that enhanced IL-5 and IL-13 secretion in an autocrine dependent manner [64]. However, regardless of whether CD4 positive Th2 or Th9 cells or ILC2 are the source, the role of IL-9 associated with EoE disease pathogenesis needs to be addressed. Clinical trials have not yet started to target IL-9 in EoE patients. Since TGF-β and IL-4 together can initiate IL-9 generation [535455], it will be interesting whether these 2 cytokines, generated by esophageal mast cells and CD4 positive Th2 cells, respectively, may promote IL-9 secretion from neighboring eosinophils, which then cause mast cells to release TGF-β and other inflammatory mediators amplifying the pathogenesis of EoE.
NKT CELLS IN EoE
Elevated iNKT cell level expressions have been implicated in EoE subjects [1821]. Esophageal tissue from patients with EoE display elevated expression of iNKT-cell-associated cell marker Vα24-Vα24Jα18positive T cells (a cell marker for iNKT cells), chemokine ligand 16 (CXCL16), and CD1d as compared to healthy control esophageal tissue [65]. This upregulated gene expression is more prominent in patients under 6 years old at diagnosis and associated with the expression of eotaxins and periostin [66]. In a food allergen-induced EoE like murine model, Rajavelu et al. [21] found eotaxins and iNKT cells were crucial for EoE progression. iNKT cells react to various stimuli, initiating the production of type 2 cytokines, and driving the development of atopic diseases [18]. Interestingly, iNKTs from patients with active EoE expand more readily and are found to produce more IL-13 in response to stimulation when compared to controls [18]. This early data from children with milk allergy found milk allergen could engage the iNKT T-cell receptor (TCR) prompting initiation of iNKT proliferation and Th2 type cytokine secretion [18].
Studies report that IL-15 receptive iNKT cells are activated in EoE and promote the severity of pediatric EoE [182167]. Both IL-15 and IL-15Ra transcripts are elevated in esophageal biopsies of patients with EoE compared to controls [67]. This suggests a role IL-15 in mediating disease by activation of Th2 adaptive immunity generating IL-5 and IL-13. Finally, co-culturing primary esophageal epithelial cells with IL-15 promotes eotaxin protein expression, supporting eosinophil chemotaxis and activation [67].
IL-18 is a pleiotropic cytokine raised in a number of eosinophilic allergic diseases as well as food allergy, eczema, asthma, and colitis [686970]. IL-18 initiates the antigen-independent activation of B cells and NKT cells [71] contributing to various gastrointestinal immune-allergic diseases including celiac disease [72]. Innate immune cells secrete IL-18 [71] which activates iNKT cells without TCR engagement [73]. A recent study found that blood IL-18 and esophageal biopsy IL-18Rα positive cells are significantly elevated in food allergen skin prick test (SPT) positive, but not SPT negative EoE patients. Furthermore, IL-18 levels correlated with esophageal eosinophilia [74].
Clinical studies show that IL-18 stimulated iNKT cells produce eosinophil activation cytokines such as IL-5 and IL-13 [18216566]. Neutralization of iNKT cells also protects against EoE development in mice [65]. These results support a role for iNKT cells in EoE pathogenesis through activation by IL-15 and IL-18 and in secreting the eosinophil activation cytokines, IL-5 and IL-13 [216574].
EPITHELIAL CELL-DC INTERACTIONS IN RESPONSE TO ALLERGENS IN EoE
Whatever the endotype of EoE, the various T cells or ILCs that control tissue inflammation need to be activated. For various types of T-cell responses, this involves the antigen presenting cell (APC) allergen capture and T-cell presentation in draining lymph nodes summarized in Fig. 4. DCs have an essential role in promoting food allergen mediated Th2 adaptive immunity in mice with prior exposure to the allergens [75]. DCs subtypes are broadly classified as conventional DCs (cDCs) and plasmacytoid DCs (pDCs). Among cDCs, CD11b positive CD172 (SIRP1α) positive cDCs that rely on IRF4 transcription factor are essential and adequate to facilitate allergic sensitization and drive Th2 polarization [76], whereas CD103 positive XCR1 positive cDCs that depend on IRF8 and BATF3 transcription factor promote food tolerance [75]. pDCs likewise support food tolerance through promoting food allergen mediated Foxp3 positive Treg cell-active responses [77]. In addition, a recent study has linked tissue epithelial cell impairment through skin barrier dysfunction in supporting the activation of DC in EoE pathogenesis [78].
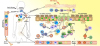 | Fig. 4Food allergen as a promoter in eosinophilic esophagitis (EoE) pathogenesis. Notwithstanding with the history of genetic disposition or gastrointestinal disease, exposure to oral food allergen would promote the epithelial barrier disruption and affect the healthy balance of body microbiota in EoE subjects. Food allergens could then infiltrate also through the skin, engage to pathogen-related receptors and drive epithelial cells activation to pro-inflammatory cytokines generation which are responsible for the trafficking of the inflammatory cells. Under epithelial injury condition, thymic stromal lymphopoietin (TSLP) generation is provoked from epithelial cells. TSLP stimulates basophils to generate interleukin (IL)-4 which support allergic sensitization after allergen being presented to a naive T cell, driving allergen-specific Th2 cells activation. Adequate allergen challenges facilitate Th2 cells recruitment and expansion, secreting vast IL-5 and IL-13 which are crucial in eosinophils trafficking and tissue remodeling. Th2 cells locally promote class switching of B cells to produce antigen-specific IgE, which binds to the surface of mast cells and produce antigen-specific IgG which engage to the surface of basophils. Activation of mast cells and basophils leads to the release of pro-inflammatory mediators such as transforming growth factor beta (TGF-β), which contributes to the local inflammatory responses and promotes remodeling and enhances muscle cell contractility. Antigen presenting cell capturing food allergens would subsequently migrate to the regional lymph nodes and thus mediating food-specific T cells activation. Food proteins may induce T-cell responses either as a consequence of antigen presentation by dendritic cells (DCs) or directly with subsequent eosinophil activation. By releasing toxic granule proteins and cytokines, eosinophils defend against invading pathogens, but cause tissue damage, stimulating fibrosis and perpetuating inflammation. In addition, fibroblasts overexpress periostin and downregulate filaggrin likely in response to IL-13. Eotaxin-3 and periostin overexpression cooperatively chemoattract CCR3 positive cells, a process primed by IL-5 which regulates eosinophil responsiveness to eotaxins and the circulating level of eosinophils. The pathomechanisms of EoE overlapping with multiple inflammatory cells are indicated. TNF, tumor necrosis factor; NKT, natural killer T.
|
There is an abundance of activated DCs in human and murine EoE tissue forming clusters with activated T cells around the esophagus and blood vessels [45]. The monocyte-derived DCs that aggregate in the esophagus have some macrophage features, such as Fc receptors CD64 and FcεRI receptors. They additionally express chitinase-like protein Ym1 which subsequently mediated macrophage activation [7980]. Their primary role is effector T-cell recruitment through chemokine production and promoting chronic esophageal inflammation [80]. Epithelial cells activate incoming monocytes by initiating chemokine and cytokine production leading to eosinophil and other innate immune cell activation. Epithelial cells go through repeated injury and repair cycles of, similar to skin lesions in atopic dermatitis (AD), contributing substantially to the process of tissue remodeling via release of repair cytokines [78].
There is increasing evidence that EoE is associated with epithelial barrier dysfunction followed by an eosinophilic inflammation similar to AD which is found in over half of patients with EoE. Epidermal differentiation complex gene expression as well as filaggrin, SPRR3 and keratins, showed a down-regulated expression in a response to IL-13 produced by esophageal epithelial cells of active EoE, which is only partially stabilized upon therapy [81]. Desmoglein (DSG)-1, an intercellular adhesion molecule responsible for epithelial integrity and barrier function was one of the most strongly downregulated genes in EoE [82]. A downregulation of DSG-1 gene, such as IL-13, caused the separation of epithelial cells (spongiosis) followed by impaired barrier function as well as by periostin induction further potentiating inflammation [82]. Ultrastructural analysis revealed a significantly decreased number of desmosomes per cell in EoE biopsies as compared to healthy controls, which was reversible after treatment [83]. Furthermore, the expression of filaggrin and the tight junction proteins zonula occludens-3 and claudin-1 is decreased in EoE, correlating with spongiosis [84]. Consistent with this finding, mutations in filaggrin are overrepresented in patients with EoE [85] and homozygous mutations of DSG1 cause a severe atopic syndrome which includes EoE [86].
Desmosomal protein cleavage and barrier integrity loss allow infiltration of allergens and organisms as well as subsequent production of danger signals and protease activated receptor (PAR)-2 activation [87]. In addition, study also found a significantly decreased protease inhibitor LEKTI expression in active EoE [40]. Evidence for this close pathogenetic link between EoE and AD is supported by the significant degree of shared genetic associations that exists among patients with these allergic syndromes, including the overabundance of genetic variants in the TSLP [81] and filaggrin loci [8285]. For example, skin injury might lead to upregulation of TSLP cytokine release which then, influences the ability of DCs to polarize T cells. Upon stimulation with TSLP, eosinophils bearing TSLP surface receptor produce extracellular DNA traps in association with eosinophil cationic proteins that are able to counteract pathogens [3358]. Interestingly, the expression of TSLP is elevated in EoE and correlated with the number of eosinophils producing eosinophil extracellular traps [40].
TSLP produced by epithelial cells in response to PAR-2, Toll-like receptor stimulation or mechanical injury, strongly induces Th2 immune responses by stimulating DCs, T cells, eosinophils, mast cells and basophils [3358]. The activation of epithelial cytokines driven by allergens supports Th2 polarization through initiating CD11b positive cDCs activation and, thus, facilitating their migration and costimulatory molecule upregulation as well as OX40L [40527576]. In addition, these activated epithelial cells also stimulate ILC2 cells and basophil activation which play a crucial role in generating IL-4 and IL-13 for driving Th2 polarization and inhibiting the development of food tolerance [33]. Besides, the proteolytic injury induces the release of IL-33, which in turn activates ILC2 cells to induce esophageal tissue eosinophilia [3978]. There are some data that ILC2 cells can be activated after allergen exposure and that their generation of IL-5 and IL-13 can affect some features of eosinophilic esophagitis in a T-cell-dependent way. Furthermore, the esophageal specific gene calpain (CAPN) 14, a member of the calpain protease family involved in the cleavage of inflammatory mediators, for instance IL-33, was upregulated in active EoE, whereas the calpain inhibitor was downregulated [88]. Consistent with these findings, hereditary variations in the CAPN14 gene locus are associated with EoE susceptibility [89] and elevation of innate cytokines, such as IL-33, has been found in epithelial cells of EoE [78].
IgE-IgG EFFECTS ON MAST CELL, BASOPHIL, OR DCs IN EoE
IgE has the lowest concentration of all antibodies, and IgE is mainly known for its role in allergic disease, where it facilitates mast cell and basophil activation when bound to the high-affinity IgE receptor FcεRI. This receptor complex is composed of FcεRIα and FcεRIβ and a dimer of FcεRIγ chains (FcεRI(αβγ2)) expressed in mast cells and basophils. The expression of FcεRI is heavily influenced by serum concentrations of IgE and also by the Th2 cytokine IL-4. Independent of atopic history, studies have also found the elevation of both B cell-mediated IgE class switching and B cell-mediated IgE-bound mast cells in EoE patients [1227]. However, there also appears to be B cell independent and IgE-independent EoE phenotypes of esophageal eosinophilic inflammation [3133]. All of these findings suggest the role of B cells and IgE in EoE pathogenesis still remains to be confirmed.
The majority of EoE patients have convincing evidence of IgE-mediated hypersensitivity to foods, as determined by elevated food-specific IgE or positive SPT [6789], despite studies showing food-triggered anaphylaxis occurring only in around 15% of these EoE patients [311]. Food allergy accounts for 19%–73% of children and 13%–25% of adults with EoE [29]. The cause for lower rates of food allergy in adults is uncertain, but this feature may imply that adults are expected to develop food tolerance [10]. Mechanistically, studies show that IgE-bearing mast cells are elevated in esophageal mucosal biopsies of EoE patients, predominantly in those that are atopic [90]. However, another study showed that IgE-mediated food allergy and EoE seem to occur independently [91].
There is local immunoglobulin class switching and IgE in the esophageal biopsies of EoE pediatric patients [1191]. However, food elimination diets, as well as anti-IgE therapy failed to confirm an IgE-mediated mechanism [9192]. A 12-week open-label single-arm study of anti-IgE antibody treatment for pediatric and adult patients with EoE in only resulted in a 33% remission rate, despite effective reduction of IgE levels in esophageal tissue [93]. Furthermore, patients with milk-induced EoE do not outgrow their disease in contrast to IgE-mediated allergy where 75% of children become tolerant by the early adolescent years [9495]. Atopic children with IgE-mediated milk allergy who undergo oral immunotherapy, are at greater risk of developing EoE once they have milk reintroduction [9194]. This is supported by the finding that higher serum specific IgG4 to food allergen, related to food tolerance rather than food allergy, has been observed in active EoE patients [25]. Clinically, patients showed the improvement on an essential elemental diet [2]. Children with food allergies and EoE frequently have a combination of findings on allergy testing, such as positive skin prick testing and patch skin testing to common food allergens, which points to a role for both IgE-mediated and cell-mediated immune response [2694]. In summary, food elimination diets based solely on IgE testing are mostly ineffective in EoE treatment [892].
The presence of allergen-specific IgE in the serum of food allergic patients with EoE leads to the suspicion that basophils and mast cells, armed with high-affinity IgE receptors, have a pivotal role in the disease pathogenesis [10112591]. FcεRI-positive cells have been observed in high numbers in the esophageal mucosa of EoE patients [96]. In addition, high expression of histamine receptors 1 and 4 in epithelial eosinophils and histamine receptor 2 in inflamed esophageal biopsies have been detected in active EoE [97], suggesting an important role of mast cells. Mast cells can also infiltrate and accumulate in the esophageal mucosa, facilitating esophageal remodeling. This likely driven by the mast cell growth factor IL-9 and triggering mast cell secretion of TGF-β and other inflammatory mediators [556190].
With respect to basophils, elevated numbers have been observed in the blood and mucosal tissues of EoE patients and also in a murine model of food allergen epicutaneous sensitization followed by oral allergen challenge [32]. Basophils are a vital source of IL-4 that could contribute to Th2-type sensitization by acting together with DCs [3398]. Cooperation between IL-33, activated basophils and ILC2 cells with basophil-derived IL-4 bolstering ILC2 cell differentiation and activation has been found in response to oral allergen challenge [33] (Fig. 4). Specifically, some key allergic signals as well as TSLP appear to initiate basophil activation in response to allergen exposures by homing to sites of inflamed tissue, promoting EoE allergic inflammatory response [99]. As discussed previously, both murine models and human studies suggest a role for TSLP and basophils in EoE [33]. Basophils can also generate lipid mediators and cytokines that lead to vessel wall extravasation and stimulate CD4 positive effector cells [99], as well as participate in tissue remodeling.
The anti-IgE antibody omalizumab has been studied in EoE with one open-labelled, single-arm study showing benefit while another study had no impact [94]. Two other studies have also shown equivocal results [98100]. These studies suggest that IgE is unlikely to have a major role in EoE pathogenesis and omalizumab is not a viable treatment strategy for EoE.
CONCLUSION
EoE is a chronic inflammatory disease with various aspects of innate and adaptive immunity to allergens, environmental triggers or viruses involved in allergic sensitization, symptoms, exacerbations, and response to therapies. There is broad crosstalk between the esophageal epithelia and immune system cells in the initiation and persistence of disease. The results from the first intervention trials in humans with EoE reflect the heterogeneity of this disease. No single drug will be effective for all patients, but some drugs might be very effective in certain patients selected by careful immunological phenotyping.
 XML Download
XML Download