

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Leiomyosarcomas originate in smooth muscle cells and account for approximately 7% of soft tissue sarcomas.1 Most of these tumors form in the retroperitoneum and intra-abdominal space. Primary leiomyosarcomas of the bone are rare, but when they do occur, they most commonly affect the metaphysis of the long bones, particularly the distal femur and proximal tibia.2
Primary leiomyosarcomas of the vertebrae are extremely rare; only 14 cases have been reported in the literature to date.3 To accurately diagnose a primary vertebral leiomyosarcoma, a metastasis from other primary sites needs to be ruled out through a systemic evaluation.
The authors encountered a very rare case of synchronous primary leiomyosarcoma in the thoracic vertebra and liver that was accompanied by advanced gastric adenocarcinoma. An alternative diagnosis of primary vertebral leiomyosarcoma of the spine with a hepatic metastasis was also considered. Synchronous primary leiomyosarcoma in the spine and liver, and primary leiomyosarcoma with liver metastasis are extremely rare.
CASE REPORT
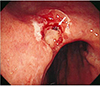
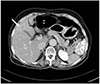
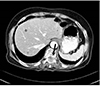
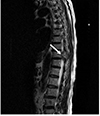
A 64-year-old woman presented to the Seoul Paik Hospital with epigastric discomfort and constipation that had lasted for 2 months. A physical examination revealed severe tenderness around the thoraco-lumbar junction. A neurological examination revealed hypoesthesia on the T10 dermatome. Her vital signs and laboratory findings were within the normal limits. Two months earlier, the patient had experienced progressively worsening lower back pain and gait disturbance, and was examined by a neurosurgeon. At that time, she was diagnosed with spinal stenosis and was prescribed nonsteroidal anti-inflammatory drugs for pain control. When she visited the department of internal medicine, an esophagogastroduodenoscopy was performed, which revealed an ulceroinfiltrative lesion measuring 1×1 cm on the gastric angle (Fig. 1). An EUS showed a heterogeneous hypoechoic mass involving all layers of the stomach wall. A biopsy of the mass confirmed a well to moderately differentiated adenocarcinoma. An abdominopelvic CT scan was performed for staging the gastric cancer, and it exposed two low attenuated lesions at the S4 and S8 regions of the liver, measuring 2.2 cm and 1.4 cm, respectively. These nodules exhibited low attenuation with rim enhancement (Fig. 2). A CT scan revealed a 3.5 cm sized soft tissue mass at the T10 vertebral pedicle and lamina compressing the dural sac (Fig. 3). Percutaneous ultrasonography-guided needle biopsy of the liver nodules showed a mesenchymal spindle cell neoplasm with diffuse smooth muscle differentiation. The neoplastic cells showed moderate to high cellularity with considerable pleomorphism (mitotic activity: 3–4/25 high power field), no identifiable atypical mitotic figures, moderate nuclear atypia, and high nuclear proliferating activity (>10% Ki-67 labelling index) (Fig. 4A). Immunohistochemical analyses revealed positive results for vimentin, neuron specific enolase, smooth muscle actin, and desmin, while the results for S100 protein, CD117 (c-kit), CD34, CD68, and MyoD1 were negative (Fig. 4B). These findings strengthened the evidence for a diagnosis of leiomyosarcoma, excluding the possibilities of a gastrointestinal stromal tumor, fibro histiocytoma, and rhabdomyosarcoma. During this process, the patient complained of severe back pain and gait disturbance that worsened progressively. MRI of the thoracolumbar spine revealed a lobulated mass compressing the spinal cord at the T10 level (Fig. 5). An emergency operation to decompress the spinal cord was performed by a laminectomy and a resection of the T9–11 spinous processes. The tumor mass was resected completely and transpedicular screws were inserted into the T9 to T11 pedicles. The histology findings from the vertebral tumor were consistent with leiomyosarcoma, which is compatible with the previous biopsy of the specimen from the liver (Fig. 4C, D). Capsule endoscopy and colonoscopy were performed to determine if there were additional primary lesions. No mass lesions were found in the small bowel lumen. Colonoscopy revealed a 0.6 cm sized Yamada type II polyp on the rectum, which was a tubular adenoma with high-grade dysplasia and focal adenocarcinomatous transformation after the polypectomy.
The potential primary cancer sites, including lung, thyroid, breast, kidney, and genitourinary organs were investigated, but there were no abnormal findings that could identify the origin of the tumor. The patient was finally diagnosed with a synchronous primary leiomyosarcoma in the spine and liver accompanied by advanced gastric adenocarcinoma, which were found incidentally, as well as a rectal polyp that demonstrated focal cancer changes. The possibilities of primary leiomyosarcoma of the spinal vertebra with hepatic metastasis and primary hepatic leiomyosarcoma with bone metastasis were also considered.
The patient refused surgery for stomach cancer. The lower back pain and gait disturbance were improved after palliative radiation therapy at the T9 to T11 vertebral levels. The patient was then referred to an oncologist for palliative chemotherapy. She received chemotherapy every three weeks with dacarbazine 200 mg/m2 and doxorubicin 15 mg/m2. After three cycles of chemotherapy, a CT scan showed that the hepatic metastatic nodules had grown. At this point, a second line of chemotherapy consisting of cisplatin 25 mg/m2 and ifosfamide 1,000 mg/m2 was administered every 3 weeks to treat the progressed metastatic leiomyosarcoma. After two cycles of this regimen, the patient decided against further chemotherapy. She died 3 months later.
DISCUSSION
Leiomyosarcoma originates predominantly in the uterus, gastrointestinal tract, skin, and soft tissues, and can metastasize to the lung, liver, kidney, brain, skin, and bone. On the other hand, primary leiomyosarcoma of bone is quite rare.3 Mirra4 reported an incidence pathologically confirmed primary bone tumors of less than 0.1%. Leiomyosarcoma commonly affects the metaphyseal portion of the long tubular bones, particularly bones near the knee joint.1 Only 14 cases of primary vertebral leiomyosarcoma have been reported thus far.3 Radiologically, leiomyosarcoma appears most often as a poorly defined osteolytic lesion with indistinct margins, moth-eaten or permeable osteolytic patterns, cortical breakthrough, and lacking a sclerotic rim,567 but these features are not specific to leiomyosarcoma. Therefore, it is necessary to distinguish it from other sarcomas, including fibrosarcoma, malignant fibrous histiocytoma, and malignant peripheral nerve sheath tumors.
A diagnosis of leiomyosarcoma is based on the electron microscopy findings and immunohistochemical staining that differentiate it from other sarcomas. Electron microscopy appears to be the most useful method. The typical electron-microscopy features of leiomyosarcoma are elongated tumor cells separated partly by collagen, a thin cytoplasmic filament that forms dense condensation, and pinocytotic vesicles.28 A histology examination revealed spindle-shaped tumor cells with eosinophilic cytoplasm and blunt cigar-shaped nuclei. The cells were arranged in parallel bundles that intersected at right angles. Immunohistochemical techniques are helpful for establishing a diagnosis because they demonstrate the origin of the smooth muscle cells. Smooth muscle actin, vimentin, and common muscle actin are moderately to highly positive in leiomyosarcoma. Desmin can be useful, but it is found in only 50% of cases,9 whereas h-caldesmon appears to be more specific.10 The present case exhibited the typical histology features and immunohistochemical stains of leiomyosarcomas, making an electron microscopic study unnecessary. The patient also had masses in the thoracic spine and liver. The findings for these two pathology specimens were ultimately found to be consistent with those of leiomyosarcoma after being stained for vimentin, smooth muscle actin, and desmin. The S100 protein staining was negative.
Although there could some debate, this leiomyosarcoma was considered to be a primary bone tumor. The primary origin of leiomyosarcoma is usually decided by the lack of a prior history of tumors and the absence of other tumors in a systemic examination using a number of measures: chest CT, abdominal CT, esophagogastroduodenoscopy, colonoscopy, capsule endoscopy, and whole-body bone scintigraphy. Generally, the origin of primary leiomyosarcoma includes vascular smooth muscle cells, multipotent mesenchymal stem cells, and intermediate cellular forms, such as myofibroblasts capable of smooth muscle differentiation.1511 Approximately 25% of leiomyosarcomas in the peripheral soft tissue may have a vascular origin. In the present case, the primary site of the leiomyosarcoma was either in the vascular tissue of the bone or in uncommitted mesenchymal cells. Whether the leiomyosarcoma in this patient originated from the spine or liver is controversial. A search for further evidence to help distinguish a primary from secondary lesion can be made. First, primary lesions tend to be relatively larger at the time of diagnosis than metastatic lesions.1213 Second, metastatic tumors tend to present as multiple lesions.14 Third, metastatic leiomyosarcomas develop more slowly in the spine and are extremely rare.15 In the present case, the size of the T-spine leiomyosarcoma was larger than that of the hepatic leiomyosarcomas. In addition, multiple lesions were found in the liver. Finally, the patient presented with spinal cord compression symptoms at the time of the initial examination. These findings led to the conclusion that the leiomyosarcomas could have originated from the thoracic vertebrae. Competing possibilities, including synchronous primary leiomyosarcomas in the spine and liver, were also considered. The chance of it being a primary leiomyosarcoma in the liver with a spinal metastasis was also considered because there was no definite evidence of a primary leiomyosarcoma in the spine with a liver metastasis, even after repeated examination of the biopsy specimens.
The clinical course of leiomyosarcoma is variable and depends on the site, extent, and resectability of the tumor, as well as the presence of metastases. Regardless of whether the tumor is primary or secondary, the life expectancy of patients with leiomyosarcomas involving the spine is 6 to 8 months.16 The recommended treatment is a surgical resection with a wide tumor free margin, but this is difficult to perform in the spine.10 If a complete resection is not feasible, radiation therapy, local tumor resection or chemotherapy can be used as a palliative treatment. If the patient has neurological symptoms, surgical decompression and reconstruction should be performed to prevent further neurological damage and functional impairment. Even when complete removal of the mass from the spine is not possible, resection of the malignant spinal tumor often reduces the neurological symptoms, relieves pain, and increases the quality of life.17 Radiation therapy for soft tissue sarcoma (STS) is applicable in several settings, including neoadjuvant, adjuvant, definite, and palliative care settings.18 Whether as a single agent or as part of a combination regimen, doxorubicin and ifosfamide have been the mainstays of systemic chemotherapy in various STSs. Dose intensive combination therapy with these agents has been found to be effective, with an overall response rate of up to 69% (95% CI, 41–89%).19 Gemcitabine and docetaxel have also been considered as active agents for doxorubicin and/or ifosfamide refractory patients with STS.20 In conclusion, this paper reported a very rare case of simultaneous primary leiomyosarcomas in the spine and liver, which was treated with a surgical thoracic spinal resection and systemic chemotherapy.

 XML Download
XML Download