

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Primary and nosocomial bacterial infections are significant causes of morbidity and mortality worldwide [1]. Identification of bacterial isolates at the species level is the first and crucial step in routine clinical laboratories, as it provides essential guidance regarding treatment. Although conventional biochemical testing is still used, whole-cell matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS) has been widely adopted for the routine identification of pathogenic bacteria [2]. MALDI-TOF MS can rapidly identify isolates by comparing the proteomic profiles of highly conserved and abundant proteins with an already compiled profile database of reference strains. Therefore, the accuracy of MALDI-TOF MS identification is heavily dependent on the software and spectral database/libraries [3]. MALDI-TOF MS is particularly useful for identifying frequently isolated pathogenic species because of better coverage of the spectral database. However, its ability to identify infrequently isolated species is questionable.
16S ribosomal RNA (16S rRNA) gene sequencing has been primarily used for cases, in which routine, conventional methods fail to identify the isolates. The 16S rRNA gene is a phylogenetic marker that is present in all bacteria and has played an essential role in the development of bacterial phylogeny and classification [45]. Recently, the similarity cut-off for 16s rRNA gene sequence (98.7%) was proposed as a boundary for bacterial species [6]. However, a 16S rRNA gene sequence similarity of ≥98.7% does not guarantee that the test isolate is a member of the species. Almost identical 16S rRNA gene sequences have been found in different species [67].
Unlike 16S rRNA gene sequencing, whole genome sequencing (WGS) provides a clear-cut criterion for bacterial classification. Furthermore, bacterial species is now defined by the relatedness of genome sequences [8]. A category of algorithms, named the overall genome-related index [4], has been devised to calculate the genomic similarity for taxonomic purposes, and a general guideline for bacterial identification using WGS data has been published [8]. WGS has considerable potential in clinical diagnostics as it could provide accurate identification of the species, and resolution can be achieved up to the strain [9]. As the cost of WGS is continuously decreasing, its use as a routine test has been validated in large hospitals [1011].
If the genome sequences of the type strains representing all the known bacterial species are available, any isolate could be identified with high confidence. However, until a few years ago, the availability of such data was not satisfactory [412]. The utility of genome-based methods for clinical diagnostics requires re-evaluation in light of the recent expansion in the bacterial genome sequence database. This is the first study to evaluate TrueBac ID (ChunLab Inc., Seoul, Korea), which is the first commercial whole genome-based bacterial identification system. Its database contains highly curated and taxonomically validated genome data of type and reference strains. We evaluated the performance of TrueBac ID in identifying clinical bacterial isolates that could not be identified using commercial MALDI-TOF MS systems and 16S rRNA gene sequencing.
METHODS
Bacterial isolates
In this retrospective study, a total of 36 clinical isolates that were either unidentified or identified with low confidence by three commercial MALDI-TOF MS systems were collected from two institutes in Korea. Fifteen isolates were chosen from Severance hospital, Seoul, Korea, and 21 isolates were from Seoul Clinical Laboratories, Yongin, Korea. The isolates were recovered from clinical specimens (blood, pus, sputum, tracheal aspirate, urine, and wounds) from April 2017 to January 2018. Since this study focuses on the identification of the isolates, an approval from the Institutional Review Board was not required, and the demographic data of the patients were not included.
Initially, we used Bruker Biotyper (Bruker Daltonics, Billerica, MA, USA) for species identification at both institutes. 16S rRNA gene PCR and sequencing were carried out for the isolates that showed no possible identification (score value: <1.70) or low confidence identification (score value: 1.70–1.99) MALDI-TOF MS results. For isolates showing uncertain identification results, we employed two additional MALDI-TOF identification systems: the VITEK MS system (bioMérieux, Marcy l'Étoile, France) and ASTA MicroIDSys (ASTA Inc., Suwon, Korea). A colony grown on sheep blood agar was smeared and dried on the target plates of each instruments. Matrix solution (α-cyano-4-hydroxycinnamic acid) and 70% formic acid (Sigma-Aldrich, St. Louis, MO, USA) provided by the manufacturer were overlaid on the spot, and the peptide profile was acquired using Microflex with Biotyper Software 3.1 (Bruker), VITEK MS V3.0 (bioMérieux), and ASTA MicroIDSys 3.0.4 (ASTA Inc.). Mass spectra were analyzed according to the manufacturers' instructions.
Genome-based identification
Genomic DNA was extracted from the isolates using the FastDNA SPIN Kit for Soil (MP Biomedicals, Santa Ana, CA, USA), and 550-bp long fragments were generated using the M220 Focused-ultrasonicator (Covaris Ltd, Brighton, UK). The sequencing library was constructed using the TruSeq DNA Library LT kit (Illumina, San Diego, CA, USA), according to the manufacturer's protocols. WGS was performed on an Illumina MiSeq system (Illumina) with a 300 bp paired-end reads sequencing kit (MiSeq Reagent Kit v3; Illumina).
The raw data from the MiSeq instrument in the FASTQ format were directly uploaded to the TrueBac ID cloud system (www.truebacid.com) and analyzed with the TrueBac ID-Genome system. The current version of the system uses trimmomatic for filtering low-quality reads [13]. The genome assembly was then carried out using the SPAdes software [14], as well as proprietary software specifically designed for the assembly of the 16S rRNA gene from the raw data.
The main section of the TrueBac ID-Genome system consists of (1) the proprietary reference database, named the TrueBac database, which is curated to hold up-to-date nomenclature, 16S rRNA gene, and genome sequences of type/reference strains, and (2) the optimized bioinformatics pipeline that provides the identification of a query genome sequence using the average nucleotide identity (ANI) [4815]. We used TrueBac database version 2018-08, which contains 10,439 genomes representing 10,152 species and 287 subspecies (7,702 with valid names, 261 with invalid names, 138 with Candidatus names [16], and 2,338 genomospecies). Genomospecies is defined as a hitherto unknown species that is supported by its genome sequences [171819]. The database also contains 18,476 16S rRNA gene sequences representing each species/subspecies.
The algorithmic identification scheme using WGS was slightly modified from that of Yoon, et al. [5]. First, the most phylogenetically closely related pool of taxa was identified using a search of three genes—16S rRNA, recA, and rplC—which were extracted from the whole genome assembly [5]. The latter two genes were a part of the 92 recently defined bacterial core genes [20]. The taxonomically meaningful similarity of 16S rRNA gene sequences was calculated as previously described [21]. In addition to the gene-based searches, we used the Mash tool (https://github.com/marbl/mash) for additional fast whole-genome based searches [22]. The top-hits of the above four searches were then pooled, and the ANI was calculated using the MUMmer tool (http://mummer.sourceforge.net/) [15].
The algorithmic cut-off for species-level identification was set at 95% ANI [815]. If the closely related taxa in a 16S rRNA gene comparison did not have the corresponding genome sequences in the database, the species assignment was made when the 16S rRNA gene sequence similarity to the best hit taxon was ≥99% with >0.8% separation between species [23]. Using these criteria, a genome sequence could be assigned to a species held in the TrueBac database, identified to the genus level (e.g. Bacillus sp.), identified as a novel species (e.g., Chryseobacterium sp. nov.), or regarded as unidentifiable.
In some cases, two or more species belonging to the same species were not yet formally reclassified. For isolates assigned to these species, the TrueBac ID system generated the final decision as a “species group” instead of individual species.
RESULTS
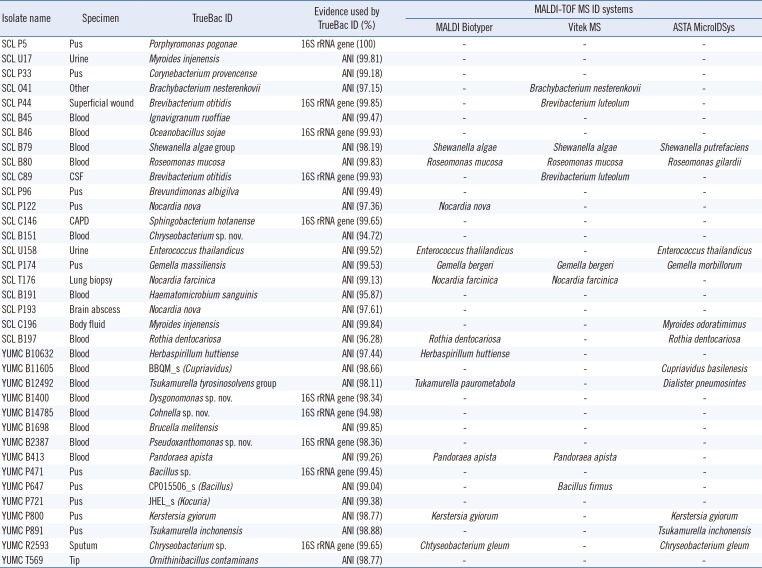
Of the 36 isolates, TrueBac ID successfully identified 25 isolates as known species (Table 1). Four isolates were new species that had not been previously recognized. Three genomospecies, labeled CP015506_s, BBQM_s, and JHEL_s, were assigned. Detailed taxonomic information on these genomospecies is available at www.ezbiocloud.net [5]. Two isolates were identified as “species group” (Shewanella algae group and Tsukamurella tyrosinosolvens group). The remaining two isolates were identified at only the genus level because of the lack of relevant reference genome sequences. Isolate YUMC P471 was most closely related to Bacillus beringensis, with 98.91% 16S rRNA gene similarity, which is lower than the cut-off (99%) we used. Isolate YUMC R2593 was found to be closely related to Chryseobacterium bernardetii and Chryseobacterium vietnamense with 99.65% and 98.92% 16S rRNA gene similarity, respectively. As the difference in 16S rRNA gene similarity between two Chryseobacterium species was only 0.73%, which is lower than the cut-off (0.8%) set for the 16S rRNA gene-based identification scheme [23], the identification was made to only the genus level. Overall, TrueBac ID identified 94% (34 of 36) of the isolates at the species level or as a novel species.
Of the 34 isolates that were conclusively identified at the species level, 26 were assigned to known species using the ANI calculation against the type strain genomes in the database, yielding a true or definitive identification. The remaining eight isolates were identified by 16S rRNA gene similarity, according to the CLSI guidelines [23].
Of the 25 isolates identified as known species by TrueBac ID, all three MALDI-TOF systems failed to identify 17 isolates. The MALDI Biotyper System identified nine (eight matched with TrueBac ID and one mismatched), the VITEK MS identified seven (three matched with TrueBac ID and four mismatched), and the ASTA system identified seven (four matched with TrueBac ID and three mismatched). The detailed identification results with the genome assembly and gene sequences we reported are available at https://www.truebacid.com/genome/demo/clinical/korea.
DISCUSSION
Overall, TrueBac ID performs well for isolates that MALDI-TOF MS systems and 16S rRNA gene sequencing fail to identify. The ability to identify rare species is largely influenced by database coverage. The TrueBac ID system contains >10,000 species, whereas commercially available MALDI-TOF MS systems contain only ~ 2,500 species [24].
Because of advances in DNA sequencing technologies and the introduction of genomics into bacterial taxonomy, numerous species have been newly described. On average, approximately 100 new species were described every month in 2017 (data from www.ezbiocloud.net). The TrueBac ID system reference database is updated every month, enabling detection of recently described species. For example, isolate SCL P33 was identified as “Corynebacterium provencense,” which was recently discovered in a human fecal specimen [25] and also associated with otitis in a cat [26]. Similarly, isolate SCL P174 was assigned to “Gemella massiliensis,” which was isolated from the sputum of a healthy individual in France [27]. Neither species could be identified by MALDI-TOF Biotyper previously [2527]. We could not identify isolate SCL P33 using any of the MALDI-TOF MS systems employed, whereas isolate SCL P174 was misidentified as Gemella bergeri using the MALDI Biotyper and VITEK MS, and as Gemella morbillorum using the ASTA system.
One of the major benefits of whole genome-based identification is that it can provide a scientifically sound decision for the recognition of novel species. We confirmed four novel species and three genomospecies based on 16S rRNA gene or genomic evidence. Isolates YUMC P721, YUMC P647, and YUMC B11605 were identified as genomospecies JHEL_s, CP015506_s, and BBQM_s, respectively. These species were never officially proposed and only tentatively named by the EzBioCloud database [5], so they can be considered novel species. As genomospecies represent previously isolated species, the use of this concept can provide further insights into species ecology. For example, the genomospecies CP015506_s included in this study is a species of the genus Bacillus, and there are three genome sequences in the EzBioCloud database from three different sources: an oral swab of a patient (USA), seawater (Korea), and soil (India) (https://www.ezbiocloud.net/genome/list?tn=CP015506_s). This additional information implies that the species is widespread in nature and may be associated with human diseases.
Isolate SCL B79 showed high ANI values to Shewanella upenei (98.19%), as well as to Shewanella algae (98.15%); both ANI values are clearly higher than the species boundary (95–96%) [8]. The ANI value between the type stains of the two Shewanella species is 98.1%, indicating that they taxonomically represent a heterotypic synonym. Similarly, isolate YUMC B12492 isolated from blood was assigned as “Tsukamurella carboxydivorans group,” which consists of Tsukamurella tyrosinosolvens and Tsukamurella carboxydivorans; these two species share high genome sequence similarity (98.9% ANI). These potential synonyms are treated as a “species group” in the TrueBac ID system to avoid possible confusion in species-level identification. We expect that all species groups we reported will eventually be combined to meet the currently accepted taxonomic scheme [8].
Overall, TrueBac ID could identify the species level for >90% of the isolates. Moreover, it demonstrated the ability to recognize new species with high confidence. This is a significant advantage of genome-based ID over other methods, including MALDI-TOF MS and biochemical tests. In addition to its superior accuracy, WGS is not influenced by media and growth conditions, in contrast to other methods based on phenotypes including MALDI-TOF MS [28].
Although 16S rRNA gene sequencing has been widely used as the gold standard for bacterial identification [29], this method is not feasible for some clinically important species with highly similar 16S rRNA gene sequences [3]. We demonstrated that WGS exhibited sufficient taxonomic coverage to be employed as a scientifically sound gold standard when any new diagnostic method or commercial system is evaluated.
This study has some limitations. We collected only those isolates that were not properly identified by MALDI-TOF MS. However, the proportion of those isolates would be low in most laboratories. In addition, the clinical significance of the isolates was not clearly defined. We assume that not all the isolates are true pathogens. Lastly, we did not examine how accurately identifying the isolates can improve patient care.
In conclusion, TrueBac ID successfully identified the majority of clinical bacterial isolates that were not identified by commercially available MALDI-TOF MS systems or 16S rRNA gene sequencing. TrueBac ID was more useful than other conventional diagnostic methods in recognizing new species. As the coverage of type strain-genome sequence database continues to grow and the cost of DNA sequencing continues to decrease, genome-based identification can be a useful tool for diagnostic laboratories, with its superior accuracy and database-driven operations.
 XML Download
XML Download