

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
As next-generation sequencing (NGS) has become more affordable and accessible globally, genetic variants have been discovered in various clinical settings. NGS is often used in the differential diagnosis of patients with cytopenias, for which MDS or another myeloid malignancy is being considered. Clonal hematopoiesis, in which hematopoietic stem cells acquire one or more somatic mutations, is more commonly recognized. In patients in whom a hematologic malignancy cannot be diagnosed definitely, the significance of clonal hematopoiesis is often unclear. This review discusses the definitions and significance of idiopathic cytopenias of undetermined significance (ICUS), clonal hematopoiesis of indeterminate potential (CHIP), and clonal cytopenias of undetermined significance (CCUS) in the context of the diagnosis and management of clonal hematopoiesis.
ICUS
In 2006, Valent, et al. [1] proposed definitions and standards to be used in the diagnosis and treatment of MDS. Originally proposed by Dr. Mufti in 2005, “ICUS” was further defined by Valent, et al. [1] as a cytopenia in one or more myeloid lineages that was persistent for six or more months, did not meet the minimal criteria for the diagnosis of MDS, and could not be explained by any other hematologic or other disease. At this time, cytopenia was defined by a Hb level of <110 g/L, an absolute neutrophil count of <1.5×109/L, or a platelet count of <100×109/L. Ten years later, these experts proposed criteria for pre-MDS conditions and updated the minimum diagnostic criteria for MDS [2]. In 2017, ICUS was defined as a cytopenia in one or more myeloid lineages that was persistent for four or more months, did not meet minimal diagnostic criteria for MDS, and again, could not be due to any other hematologic or other disease [2]. Importantly, cytopenia was defined as any decrease from institutional reference values. The clinical course of patients with ICUS varies. A subset of patients with ICUS will progress to a myeloid malignancy, including MDS and AML.
A few years after these cases of ICUS, researchers identified a phenomenon they termed “clonal mosaicism,” whereby small clones exhibiting large structural chromosomal abnormalities were identified in a small subset (<1%) of healthy individuals [345]. They noted that these abnormalities increased with age and were associated with a subsequent increased risk of hematologic cancer; however, they were also sometimes eliminated from the body without treatment.
AGE-RELATED CLONAL HEMATOPOIESIS
Large-scale genome wide association studies identified what was termed as clonal hematopoiesis in healthy populations. Jaiswal, et al. [6] and Genovese, et al. [7] simultaneously published studies involving thousands of patients presumed not to have hematologic malignancy (patients selected for mental health studies and diabetes studies and controls). They discovered that clonal genetic mutations previously associated with hematologic malignancies could be identified at low levels in a subset of healthy individuals. This trend increased with age and was termed “age-related clonal hematopoiesis.”
Not all clonal hematopoiesis cases involve known or candidate driver mutations of hematologic malignancy. Zink, et al. [8] could prove it using whole genome sequencing, a method that does not rely on the detection of these driver mutations. They showed that clonal hematopoiesis might be far more common than previously understood, even trending with age towards inevitability. Furthermore, in most cases (87.4%) of clonal hematopoiesis, known driver mutations are not present. Interestingly, regardless of whether a driver mutation was present, reduced overall survival and increased risk for hematologic malignancy were still associated with the presence of clonal hematopoiesis, indicating there is still much unknown about the pathogenesis involving these small hematopoietic clones.
CHIP
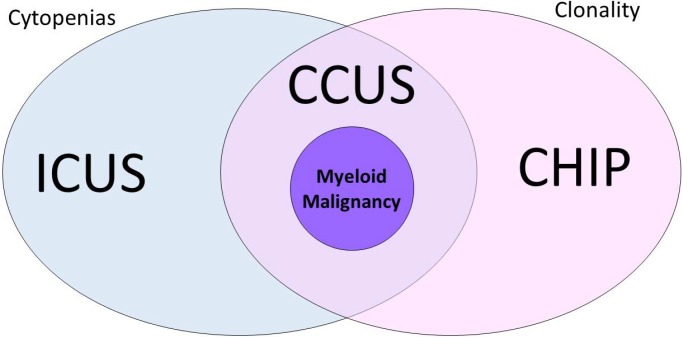
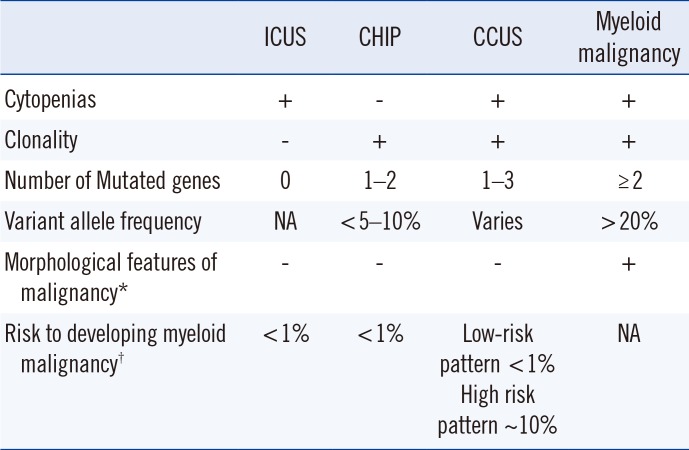
Arising from this landscape of unexplained cytopenias and clonal hematopoietic genetic mutations in the elderly, the term “CHIP” was coined [9]. CHIP is a clonal pre-malignant disorder similar to monoclonal gammopathy of undetermined significance. CHIP delineates individuals with a somatic mutation that is associated with hematologic malignancy but without fulfillment of other diagnostic criteria for a hematologic neoplasm [9]. Specifically, CHIP requires the absence of morphological evidence of a hematologic neoplasm, the absence of criteria for paroxysmal nocturnal hemoglobinuria, monoclonal B-cell lymphocytosis, and monoclonal gammopathy of undetermined significance, and the presence of a somatic mutation associated with hematologic neoplasm at a variant allele frequency (VAF) of at least 2% [9]. VAF, defined as the number of reads containing the variant of interest divided by the total number of reads, is associated with somatic variants and can also be informative [10]. Cases of cytopenia and clonal hematopoiesis are described as “CCUS.” CHIP and ICUS have some degree of overlap, and we can now define a “non-clonal” ICUS category without any identifiable mutations—a “CHIP” category with clonal mutations, but without cytopenias or symptomatology; and a third CCUS group that is essentially a subset of CHIP in patients who do not meet the criteria for MDS but who do have cytopenias (Table 1; Fig. 1).
In fact, many of the mutations found in MDS and CCUS occur in CHIP, including DNMT3A, TET2, and ASXL1 [1112]. The odds of progression to overt neoplasia with CHIP are <1% per year [9]. The detection of variants associated with CHIP increases in frequency with age [6]. However, while CHIP and MDS both affect older individuals, CHIP is far more frequent in the elderly [6913]. Importantly, Jaiswal, et al. [6] showed that the presence of clonal hematopoiesis is associated with an increased risk of all-cause mortality, including increased risks of coronary heart disease and stroke. It is now thought that CHIP results in disordered hematopoiesis through clonal monocytes/macrophages that accelerates atherogenesis and promotes cardiovascular disease in patients with other risk factors and inflammatory disorders [14]. Jaiswal, et al. [14] showed it using a mouse model, in which mice prone to hypercholesterolemia were grafted with bone marrow with a TET2 mutation via knockout. Mice engrafted with TET2-mutated allografts showed larger atherosclerotic lesions than those engrafted with normal control bone marrow allografts. They concluded that CHIP approximately doubles the risk of coronary heart disease [14]. Adverse outcomes with CHIP were also reported for clonal hematopoiesis after autologous stem cell transplantation for lymphoma [15]. It remains unclear whether other conditions are affected by clonal hematopoiesis.
More recent research has helped to stratify clonal hematopoiesis into cases that are less likely to progress (or at least not as rapidly) to an overt hematologic malignancy versus those molecular profiles where transformation to a malignant state may be more imminent [9]. A more robust understanding of these conditions will likely allow for highly granular associations between particular gene mutations, co-mutation patterns, and/or variant allele fractions with specific clinical phenotypes. The accuracy of MDS diagnosis and prognostication will thereby be improved, as will the clinicians' ability to rule out this condition with a high degree of confidence because of the high negative predictive value associated with the lack of somatic mutations in cytopenic patients [9].
CCUS
The acquisition of a clonal abnormality in patients with cytopenia(s) for whom minimal diagnostic criteria for MDS are not met and no other hematologic neoplasm is detected is termed “CCUS” [29]. Many of the somatic mutations found in MDS [1012] are in fact also found in pre-MDS patients or CCUS [1617], including mutations in TET2, SF3B1, ASXL1, SRSF2, DNMT3A, and RUNX1 [1617]. While it was initially thought that MDS-associated somatic mutations alone are not diagnostic of MDS in patients with cytopenias [18], a more recent study by Malcovati, et al. [19] has shown that in patients with minimal or no dysplasia, the presence of CCUS and a “high mutation pattern” of genes can be used to diagnose or predict a myeloid neoplasm. They found no difference in survival between patients with MDS versus CCUS with an accompanying “high mutation pattern,” a term specifically defined in their study as mutations in a spliceosome gene (SF3B1, SRSF2, U2AF1), a mutation in TET2/ASXL1/DNMT3A, and one or more other mutation(s). The presence of CCUS with one or more mutation(s) carries a 10-fold risk for development of a myeloid neoplasm versus ICUS with no mutations. A VAF >10% is also a relevant threshold as Malcovati, et al. [19] applied a 10% VAF filter in studying associations between mutation pattern and diagnosis. Conversely, the absence of mutations or mutations in less specific genes (e.g., DNMT3A alone) does not exclude the diagnosis of a myeloid neoplasm [19]. Thus, the presence of myeloid gene mutations, number of mutations, mutational burden, and pattern of mutations have prognostic value.
Specific gene mutations or patterns of co-mutation correlate with clinical phenotypes and thereby confer a higher positive predictive value for specific diagnosis of myeloid neoplasm. Co-mutation of TET2 with ASXL1 or spliceosome genes SRSF2 or ZRSR2 is characteristic of chronic myelomonocytic leukemia [11]. Mutation of the spliceosome gene SF3B1 (particularly Lys700Glu) is closely associated with ring sideroblasts in MDS [20]. Malcovati, et al. [19] showed that co-mutation of DNMT3A, TET2, or ASXL1 (CHIP-related mutations) with a spliceosome gene (e.g., U2AF1, SRSF2, EZH2, SF3B1, or ZRSR2) is especially closely related to myelodysplasia and carries a positive predictive value of 86–100%, despite not being diagnostic of a myeloid neoplasm without relevant WHO-defined criteria. Therefore, while spliceosome mutations do occur in the context of CHIP, specific patterns involving these gene mutations carry strong predictive value for the diagnosis of a myeloid neoplasm. These data support the clinical utility of mutation profiling by NGS on peripheral blood or bone marrow samples from patients with unexplained cytopenia(s). Detection of these mutation patterns in cytopenic patients should increase the diagnostic yield and improve overall diagnostic accuracy.
Some mutations, especially those widely found in age-related clonal hematopoiesis, as well as in myeloid malignancies, have shown varying impact in different studies. DNMT3A is a good example of this; in one study that examined AML patients with prior evidence of lympho-myeloid clonal hematopoiesis (DNMT3A mutations present in both myeloid and T-cells), the majority of patients had a pre-leukemic clone that was refractory to chemotherapy and also represented the founding clone at relapse [21]. Conversely, a recent study found that DNMT3A clonal mutations are vastly more prevalent than other mutations (along with TET2) in hematologically normal individuals [22]. Based on methodology with increased sensitivity, they appear to be two- to three-fold more prevalent in the general population than previously thought; interestingly, DNMT3A mutations showed no impact on blood counts, with neither cytopenias nor proliferative effects produced by these clones. This study also failed to find a cardiovascular risk association, as previously reported [14]. As for oncogenic potential, the authors argue that with the abundance of DNMT3A variants in the aging population, these “pre-leukemic” clones are more a matter of coincidence than true oncogenic potential [22].
Clonal hematopoiesis is clearly a complex process and appears to represent more than a possible pre-malignant state. Interestingly, in patients with dyskeratosis congenita (a bone marrow failure syndrome), increased clonal hematopoiesis was observed in some patients with improved or restored hematopoiesis [23]. However, these may not be the same driver mutations typically associated with an increased risk of hematologic malignancy. Aplastic anemia patients show enrichment for clonal hematopoiesis (identified in nearly half of patients in one study), with approximately one-third of patients specifically showing somatic mutations in myeloid malignancy candidate genes, although these were usually at low allelic burdens of <10% [24].
One issue that is becoming increasingly apparent as data continue to accumulate is that the identification of a somatic mutation representing a clone is not a “one size fits all” finding. Some clonal mutations appear more benign than others, and different genes and mutations are associated with different findings. For example, in aplastic anemia, DNMT3A and ASXL1 clones tended to be age-related, to increase over time, and to be associated with a worse outcome, while BCOR, BCORL1, and PIGA mutations correlated with a better response to immunotherapy and improved survival outcomes [24]. Profiles of somatic mutations in aplastic anemia could potentially be used in a similar way to evaluate patients for MDS; certain profiles are likely to herald transformation to MDS or AML and might warrant early intervention [25].
Most (approximately 95%) individuals with CHIP demonstrate a single somatic variant with a minority (5%) showing variants in two different genes. Exceedingly few individuals with CHIP show more than two distinct variants, a finding with potential diagnostic utility in the setting of myeloid neoplasms. Three of the most commonly mutated genes in individuals with CHIP, as well as more broadly in myeloid malignancies, are DNMT3A, TET2, and ASXL1 (Table 2) [1226]. These genes are generally involved in DNA methylation and epigenetic modification, mutation of which may confer a growth advantage, which drives clonal cell expansion [2728]. CHIP may also involve splicing factors, such as SF3B1, SRSF2, U2AF1, and ZRSR2 [19]. Surprisingly, several other genes are less frequently mutated in CHIP, including TP53 and JAK2, as well as BCOR, BCORL1, CBL, and PPM1D. In CHIP, VAF may show a range; however, VAF significantly >10% is uncommon in CHIP and should raise suspicion of an underlying hematologic malignancy [17].
MANAGEMENT
Clearly, clonal hematopoiesis plays a significant role in both asymptomatic patients and those with cytopenias. These potential outcomes raise concerns regarding diagnostic strategies and whether large-scale screening of patients should be conducted. As CHIP is present in 10–15% of patients >70 years old, the clinical management of such patients also poses significant challenges. Management of such patients with CCUS includes monitoring disease progression and consideration of cardiovascular risk factors [29]. For patients with CHIP alone (i.e., without cytopenias), management should focus on cardiovascular risk factors, given the increased risk of stroke and coronary heart disease [30].
The clinical utility of targeted NGS panels that provide comprehensive assessment of CHIP-related mutations is increasingly apparent in the context of cytopenic patients, for whom a diagnosis of MDS is being considered [3132]. While further studies relating somatic mutations to clinical impact are necessary, accumulating data may facilitate the designation of “MDS-associated somatic mutations” (or patterns of co-mutation) enabling a definitive diagnosis of MDS in the absence of morphological criteria, similar to the “MDS-associated cytogenetic abnormalities” established in the current WHO classification for MDS [33]. Numerous somatic mutations also carry prognostic value in MDS; however, they are yet to be incorporated into existing prognostic scoring systems [3435]. This contrasts with age-related clonal hematopoiesis in the absence of cytopenias, where the concern is not hematologic malignancy, but rather a cardiovascular risk factor. Further understanding of the pathways driving expansion of clonal hematopoiesis and its interaction with atherogenesis may lead to better intervention strategies.

 XML Download
XML Download