

 PDF
PDF ePub
ePub Citation
Citation Print
Print



Dear Editor:
Hyaline fibromatosis syndrome (HFS) is a rare genetic disease inherited in autosomal recessive pattern with germline mutations of gene encoding anthrax toxin receptor-2 (ANTXR2). HFS affects skin, joints, bones and internal organs due to abnormal accumulation of hyaline substances12. The main clinical features include papular skin lesions, hyperpigmentation over the joints, limitation of movement, recurrent infection, gastrointestinal symptoms and failure to thrive3.
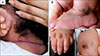
A 1-year-old male patient from the United Arab Emirates visited for multiple pearl-like papules on head and neck, which had started 3 months ago (Fig. 1A). There was hyperpigmentation over malleolus, joint of hand and knee (Fig. 1B). In addition, vomiting, diarrhea, microcephaly, muscular rigidity and joint contracture were seen. The patient was a first child and born to healthy 1st cousin consanguineous parents.
There were osteopenia and cortical depression in tibial metadiaphysis in plain radiograph which might be associated with metabolic bone disease or bony dysplasia and rare connective tissue disease. Genetic analysis revealed a homozygotic mutation in the 710th nucleotide of the coding region of the ANTXR2 gene in q-arm of chromosome 4. The nucleotide was changed from thymine to guanine and the protein was changed from isoleucine to serine (Fig. 2). The patient was diagnosed as HFS and this gene mutation was a new variant.
HFS is a rare genetic disorder caused by a mutation in ANTXR2 gene. There are two subtypes. Infantile systemic hyalinosis, a severe form, causes fatal symptoms and death before first 2 years of life due to hyaline substances deposition in multiple organs and recurrent infection. Another type, called juvenile hyaline fibromatosis, is a mild form with less severe symptoms. Our patient was thought to belong to infantile systemic hyalinosis because he had systemic involvement including skin, muscles and bones before 1 year of age. Recently, researchers believe that these two types are in the same disease spectrum called HFS2. Patients are usually normal at birth and abnormalities begin in the first few months. Hyperpigmentation occurs over the joints with thickening of the skin. Pearl-like papules are around the face and neck, and masses are around the anus. Progressive joint contractures occur. HFS can cause gastrointestinal problems, gingival hypertrophy, and life-threatening complications.
Pathogenesis is not yet clear, but there are several hypotheses. ANTXR2 gene encodes proteins involved in the formation of tiny blood vessels. Some researchers believe that this protein is also important for maintaining the basement membrane. Mutation of ANTXR2 gene leads to malformation of the basement membrane and it is presumed that the hyaline substances leaks out and accumulates in tissue and causes HFS4.
In conclusion, HFS is rarely reported less than one hundred cases5 and caused by the mutation in the ANTXR2 gene. HFS is mainly reported in Arab countries and our patient is also from Arab countries. Since the disease is inherited in autosomal recessive pattern, the disease usually develops among consanguineous families. Herein, we report a rare case of HFS with a new variant of genetic mutation in previously unreported location of ANTXR2 gene.

 XML Download
XML Download