

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Hereditary sensory and autonomic neuropathy type IV (HSAN-IV), Gitelman syndrome, and Wilson disease are all rare autosomal recessive genetic disorders. HSAN-IV is characterized by congenital insensitivity to pain, anhidrosis, inability to sweat, leading to defective thermoregulation, developmental delay, and self-mutilation [1]. It is caused by mutations in neurotrophic receptor tyrosine kinase 1 (NTRK1) gene which encodes high-affinity tyrosine kinase receptor [2]. No specific treatments have been established for HSAN-IV. Thus, a multidisciplinary approach is required for its treatment [3].
Wilson disease can have various clinical manifestations, ranging from asymptomatic hepatomegaly to fulminant hepatitis, acute pancreatitis, neurological disorder, and hemolytic anemia [456]. Wilson disease is a rare, inherited autosomal recessive disease caused by mutations of ATPase copper transporting beta (ATP7B) gene, leading to excessive accumulation of copper in specific organs such as the liver and brain [7].
Gitelman syndrome is also a rare genetic disorder with kidney involvement. It manifests as hypokalemia, hypomagnesemia, and metabolic alkalosis [8]. Gitelman syndrome is caused by mutations in solute carrier family 12 member 3 (SLC12A3) gene [9], leading to loss of function of sodium channel transport in distal convoluted tubule [10].
The possibility of three concurrent autosomal recessive disorders developing in the same patient may be extremely rare. Here we report a case of Wilson disease manifested as fulminant hepatitis, acute pancreatitis, and acute kidney injury in a 15-year-old boy with comorbidities of HSAN-IV and Gitelman syndrome.
CASE REPORT
A 15-year-old boy visited the emergency department presenting complaint of vomiting and lethargy. He was admitted to the general ward at Seoul National University Bundang Hospital. Two weeks before admission, he had experienced left thigh swelling after falling from a bed at a private rehabilitation center. Radiologic examination at the emergency department revealed a left femur shaft fracture. Two days later, he underwent orthopedic surgery for the femur fracture. He was discharged from the hospital without any complications seven days after surgery.
Two days before the present admission, he suddenly began to have poor oral intake. The patient refused to eat solid foods. He only took in liquids. His body temperature was elevated to 38.0°C. One day before admission, bilious vomiting developed and the patient became lethargic. In addition, his urine output decreased. It became dark in appearance. As gastrointestinal symptoms persisted with features of dehydration, he was admitted to the general ward for supportive care and further evaluation of the bilious vomiting.
As the patient was an abandoned child residing in a private rehabilitation center, his perinatal history or family history was unknown. His past medical history in infancy was unclear either. He was initially diagnosed with cerebral palsy of unknown cause. Given his pain insensitivity with anhidrosis, he was later diagnosed with HSAN-IV at Seoul National University Bundang Hospital. He had suffered from recurrent orthopedic diseases such as femur fractures, septic arthritis, and osteomyelitis approximately 1–2 times per year. His fingertips were mutilated because of recurrent cellulitis and septic arthritis of fingers. He had recurrent trauma history due to an impulse control disorder and pain insensitivity as well as poor hygienic condition. His body temperature was unstable. It tended to change dramatically, ranging from abnormally low body temperature to high fevers along with unstable blood pressures indicative of autonomic dysregulation. He had been treated for severe dry skin, hyperkeratosis, fissuring, and xerotic eczema with topical ointments. He was wheelchair-bound and mentally retarded, although verbal communication with social workers was partially possible. During long-term follow-up at the Pediatric Nephrology Clinic of the same hospital, hypokalemia and hypomagnesemia were also detected, leading to suspicion of Gitelman syndrome. With regard to medications, he was taking anti-psychotics, psychostimulants, and spironolactone.
On physical examination, he was alert but cachectic. He was wearing a hip spica cast for the femur fracture. His height was 134 cm (<3rd percentile). His body weight was 29.1 kg (<3rd percentile). He had blood pressure of 130/88 mmHg, pulse rate of 97 beats/minute, respiratory rate of 21 times/minute, and body temperature of 37.3°C initially. Neither conjunctiva was anemic. Neither sclera was icteric. His lips and mucous membranes were slightly dehydrated. Both tympanic membranes were intact. His cervical lymph nodes were not palpable. The chest wall expanded symmetrically without retraction. Breath sounds were clear without crackles or wheezing. His heartbeat was regular without murmur. His abdomen was soft and flat. There was no tenderness or rebound tenderness in the abdomen. Bowel sounds were hyperactive. There was no shifting dullness on percussion. His liver or spleen was not palpable. There was no peripheral edema, digital clubbing, or cyanosis. However, all fingertips were mutilated (Fig. 1). His skin was dry. There was no rash or petechia.

On the day of admission, laboratory tests were done with the following results: sodium, 138 mmol/L; potassium, 3.2 mmol/L; aspartate transaminase (AST), 23 IU/L; alanine aminotransferase (ALT), 12 IU/L; total bilirubin, 1.0 mg/dL; blood urea nitrogen (BUN), 29 mg/dL; creatinine, 0.41 mg/dL; C-reactive protein (CRP) <0.03 mg/dL; and serum magnesium, 1.6 mg/dL. Supportive treatment was started to correct his dehydration and he was maintained in stable condition.
On the third day of hospitalization, a high fever developed suddenly and his temperature rose to 41.8°C. He had no response to antipyretic drugs. At the same time, his blood pressure suddenly dropped to 50/17 mmHg. He also had seizure-like movements, including upward eyeball deviation, loss of consciousness, and generalized clonic movement. Both pupils became fixed at 2 mm diameter. Results of laboratory tests were as follows: white blood cell (WBC) count of 19.4×103/µL, hemoglobin level of 9.6 g/dL, platelet count of 90×103/µL, sodium level of 137 mmol/L, potassium level of 2.6 mmol/L, AST level of 454 IU/L, ALT level of 258 IU/L, total bilirubin level of 4.3 mg/dL, direct bilirubin level of 2.6 mg/dL, amylase level of 228 IU/L, lipase level of 888 IU/L, creatinine phosphate kinase level of 1,258 IU/L, lactate dehydrogenase level of 985 IU/L, BUN level of 49 mg/dL, creatinine level of 1.62 mg/dL, and CRP level of 0.6 mg/dL. These results indicated acute liver failure, acute pancreatitis, disseminated intravascular coagulation, rhabdomyolysis, and acute kidney injury. Additional testing for anemia revealed plasma hemoglobin level of 27.5 mg/dL, serum ferritin level of 250 ng/mL, and weakly positive antinuclear antibody (1:40).
Repeated blood testing results on the same day revealed markedly worsened results as follows: AST, 492 IU/L; ALT, 545 IU/L; total bilirubin, 10.8 mg/dL; direct bilirubin, 5.1 mg/dL; amylase, 520 IU/L; lipase, 5,071 IU/L; prothrombin time international normalized ratio (PT INR), 3.1; activated partial thromboplastin time (aPTT), 52.7 seconds; fibrinogen, 160 mg/dL; D-dimer, 17.62 µg/mL; alkaline phosphatase, 126 IU/L; and γ-glutamyltranspeptidase, 98 IU/L. Aggressive management was performed, including fluid resuscitation, intravenous inotropes (dopamine and norepinephrine), antibiotics, and gabexatemesilate. He was transferred to the pediatric intensive care unit (PICU) and ventilator care was applied. In the PICU, he developed seizure-like movements. Lorazepam was injected once and mannitol was started to reduce intracranial pressure. Abdominal computed tomography revealed no definite radiological evidence of acute pancreatitis. However, multiple filling defects were noted in both kidneys. Findings of brain magnetic resonance imaging suggested hypoxic ischemic encephalopathy.
Further investigation of the etiology of fulminant hepatitis revealed a low serum level of ceruloplasmin (5.5 mg/dL) and increased 24-hour urine copper level (1,028 µg/dL). Liver biopsy was done under the suspicion of Wilson's disease. Liver pathology revealed centrilobular confluent necrosis with marked copper deposition in hepatocytes and Kupffer cells with extensive lobular disarray (Fig. 2). Liver copper deposit was 74 µg/g dry weight (reference range 10–35 µg/g). Ophthalmologic examination was negative for Kayser–Fleischer rings.
Fig. 2
Liver biopsy revealing centrilobular confluent necrosis with marked copper deposition in hepatocytes and Kuffer cells with extensive lobular disarray. (A) Copper granules (red) in periportal hepatocytes (Rhodamine stain, ×400); (B) centrilobular necrosis of the liver (hematoxylin and eosin stain, ×400).

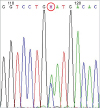
To confirm the diagnosis of suspected underlying genetic diseases based on his past medical history and present manifestations, gene studies for HSAN-IV, Gitelman syndrome, and Wilson disease were performed. HSAN-IV was confirmed based on results of NTRK1 gene sequencing for HSAN-IV which revealed c.2002G>T, p.Asp668Tyr heterozygote (American College of Medical Genetics and Genomics /Association for Molecular Pathology [ACMG/AMP] classification: pathogenic) and c.360-1G>A, (IVS3) heterozygote (ACMG/AMP classification: pathogenic), indicating a compound heterozygote mutation causing autosomal recessive disease (Fig. 3). In addition, SLC12A3 gene sequencing for Gitelman syndrome revealed a heterozygote c.1216A>C in exon 10 [p.Asn(AAT)406His(CAT)] (Fig. 4). However, ATP7B gene sequencing for Wilson disease showed negative results despite clinical manifestations, laboratory and liver pathologic findings, and a clinical course compatible with the diagnosis of this disease.
Fig. 3
Gene sequencing of NTRK1 for hereditary sensory autonomic neuropathy type IV revealing (A) c.2002G>T, p.Asp668Tyrc; (B) 360-1G>A mutation.

Fig. 4
Gene sequencing of SLC12A3 for Gitelman syndrome in the same patient showing a heterozygote c.1216A>C in exon 10 [p.Asn(AAT)406His(CAT)].
After initiating treatment with trientine (triethylenetetramine) 25 mg/kg/day via nasogastric tube as well as a low copper diet, clinical symptoms and laboratory findings began to improve markedly. Laboratory tests results on the 15th day in the hospital were as follows: WBC count of 8.66×103/µL, hemoglobin level of 12.9 g/dL, platelet count of 364×103/µL, sodium level of 137 mmol/L, potassium level of 4.0 mmol/L, AST level of 128 IU/L, ALT level of 127 IU/L, total bilirubin level of 1.3 mg/dL, BUN level of 25 mg/dL, creatinine level of 0.1 mg/dL, PT INR of 1.15, and aPTT of 52.3 seconds.
As the patient did not recover from brain damage caused by high fever and shock and his ability to breathe independently was not restored during hospitalization, he received a tracheostomy for long-term mechanical ventilator care and percutaneous endoscopic gastrostomy for enteral feeding. In a bed-ridden state on home ventilator care and gastrostomy feeding, trientine treatment and copper restriction diet were maintained without any problem after discharge from the hospital or during long-term follow-up at the outpatient clinic.
This study was approved by the Institutional Review Board (IRB) of Seoul National University Bundang Hospital (IRB no. B-1703-387-701). The need for informed consent was waived by the board.
DISCUSSION
To our knowledge, this is the first case of new-onset Wilson disease in a child with genetically confirmed HSAN-IV and Gitelman syndrome. HSAN-IV or congenital insensitivity to pain with anhidrosis is a rare autosomal recessive disorder. It is caused by mutations in the NTRK1 gene which encodes high-affinity tyrosine kinase receptor [2]. Children with HSAN-IV typically show mental retardation and a low intelligence quotient score. Most children with HSAN-IV have behavioral problems, ranging from autism to aggressive behaviors, attention deficit hyperactivity disorder, and neurodegenerative processes [111213]. HSAN-IV involves the central nervous system [14], the peripheral nervous system, musculoskeletal, endocrine, ophthalmic, oral, and immunological systems [15]. Almost 20% of patients with HSAN-IV die within the first 3 years because of hyperpyrexia [15]. Early diagnosis and early intervention are very important for the prevention and treatment of various complications [16]. The etiology and pathogenesis of the condition remain unclear. Our patient had mental retardation with attention deficit hyperactivity disorder, insensitivity to pain, leading to self-mutilation, recurrent osteomyelitis, and multiple fractures. He also suffered uncontrolled hyperpyrexia because of his inability to sweat and severe dry skin.
To date, case of HSAN-IV concurrently with Gitelman syndrome and Wilson disease has not been reported. These three diseases are all very rare genetic conditions in the general population. In the present case, the diagnosis of HSAN-IV was clinically and genetically confirmed because his clinical features and disease course were typical and gene sequencing for NTRK1 gene revealed compound heterozygote mutations of the gene [17]. Clinical manifestations of the patient and laboratory findings showed hypokalemia, hypomagnesemia, and metabolic alkalosis that were highly compatible with Gitelman syndrome [8]. Although the patient in this study was not completely confirmed to have Gitelman syndrome as gene sequencing revealed only a single heterozygote mutation of SLC12A3 gene, there might be another mutation that was not detected by Sanger sequencing.
However, genetic diagnosis of Wilson disease using Sanger sequencing in this patient failed to confirm the diagnosis despite his typical clinical features and response to treatment as well as unique laboratory and pathologic findings of Wilson disease. The diagnostic criteria for Wilson disease include the following: 1) low serum copper level of less than 1 mmol/dL or 10 mmol/L; 2) low serum ceruloplasmin level of less than 20 mg/dL or 200 mg/L; 3) excessive urine copper levels of above 1 mmol/24 hours and >100 µg/24 hours, particularly after initiating oral penicillamine treatment (usual dosage of 20 mg/kg/day); and 4) an elevated hepatic copper levels of more than 250 mg/g dry weight [46]. Our patient showed a low serum ceruloplasmin level and excessive urine copper in addition to hepatic copper dry weight that was higher than the normal range. Additionally, as soon as our patient was treated with a low copper diet and 25 mg/kg/day of trientine, the aggressive state of fulminant hepatitis began to subside and serum levels of liver enzymes and bilirubin started to normalize rapidly, with stabilization of coagulation panels, similar to those reported in previous studies [4]. Wilson disease can have a variety of clinical manifestations, such as asymptomatic hepatomegaly, subacute or chronic hepatitis, acute liver failure, acute pancreatitis, neurologic disorder, Coombs negative hemolytic anemia, renal Fanconi syndrome, and progressive renal failure [456]. In this case, his clinical symptoms, pathologic findings, laboratory findings, and treatment response were compatible with those of Wilson disease. His total score (a total score of 6) according to the Wilson disease scoring system confirmed the diagnosis to be highly likely [18].
In summary, here we report a rare case of Wilson disease that developed as acute liver failure and acute pancreatitis in a child with genetically confirmed HSAN-IV and Gitelman syndrome. These diseases are all rare autosomal recessive disorders with different gene loci. From this case, we recognize that even when there is a known underlying rare disease, other rare diseases should be considered and evaluated if they are strongly suspected on the basis of definite clinical evidence. Early detection and early treatment may improve the prognosis of the disease and augment the quality of life of the patient. If the patient in this case had been diagnosed earlier enough to preserve neurological status, he might have a better prognosis.
 XML Download
XML Download