

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Tuberous sclerosis complex (TSC) is an autosomal dominant genetic disorder characterized by hamartomatous tumors in the brain, eyes, heart, lung, skin, and kidney1). The incidence rate of TSC is 1/6,000 to 1/10,000 live births, affecting 2 million people globally123). The kidney is one of the major target organs in TSC, and renal diseases are found in up to 80% of autopsy cases4). Angiomyolipoma (AML), the most common renal disease in these patients, is frequently related to renal impairment and greatly contributes to TSC mortality due to its complications5). Although nephrologists often encounter patients with TSC and AML in clinical practice, treatment is mainly focused on preventing acute bleeding and managing increasing tumor burden6). Metabolic aspects in patients with TSC are relatively unknown. Only a few previous studies have reported that patients with TSC had deranged bone mineral metabolism7), but no such cases have been reported in Korea. Here, we report a case of a patient with TSC who presented with severe hypocalcemia.
Case Report
A 63-year-old man was admitted to the nephrology department for further evaluation of renal impairment and hypocalcemia. The patient was clinically diagnosed with TSC at another tertiary hospital approximately 40 years ago and had a history of hypertension and diabetes. He had not undergone thyroid or parathyroid gland surgery. Several episodes of seizures occurred when he was young. These were managed with anticonvulsants, which were maintained until several years ago. He had also been taking unidentified herbal medicine due to fatigue for a month. He had no cognitive or behavioral problems. He had large bilateral renal AMLs and underwent transcatheter arterial embolizations due to left and right side hemorrhages 24 years and 2 weeks ago, respectively. His two sons were also diagnosed with TSC based on genetic test results.
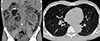
On admission, the patient complained of general weakness and muscle tremor without other neurologic symptoms, such as tetany or seizure. Physical examination revealed trace peripheral edema and facial angiofibroma only. His blood pressure was 113/67mmHg, with a pulse rate of 87/min, a respiratory rate of 18/min, and body temperature of 36.9℃. His initial laboratory findings were as follows: white blood cells, 12,760/mm3; hemoglobin, 7.9 g/dL; platelets, 365 K/mm3; blood urea nitrogen, 54 mg/dL; serum creatinine, 5.26mg/dL; alkaline phosphatase, 98 IU/L; magnesium, 2.1mg/dL; total calcium, 3.7 mg/dL (corrected calcium, 4.5mg/dL); ionized calcium, 0.591mmol/L; phosphrous, 5.0mg/dL; and albumin, 3.0 mg/dL. An electrocardiogram showed QT prolongation (QT 440 ms, QTc 508ms). Computed tomography (CT) demonstrated enlarged and distorted kidneys due to numerous AMLs and a large number of tiny nodules in both lungs, consistent with renal and pulmonary manifestations of TSC(Fig. 1). His baseline kidney function was not determined due to the lack of previous medical records; however, severely destructed renal parenchyma detected in CT suggested progression to advanced chronic kidney disease (CKD). Bone mineral density was within normal range (the lowest value was −0.6 for the total femur). To determine the cause of the hypocalcemia, hormonal status related to calcium homeostasis was examined, including parathyroid hormone (PTH) 44 pg/mL, 25-(OH) vitamin D3 9 ng/mL, and 1,25-(OH) 2 vitamin D3 2.8 pg/mL (reference range, 19.6-54.3 pg/mL). Urinary calcium concentration was very low(<0.8mg/dL), and the urinary calcium/creatinine ratio was <0.013mg/mg Cr. The fractional excretion of calcium(FECa) was 0.015.
The patient was initially treated with oral calcitriol 0.25 mg/day, calcium acetate 4.26 g/day, and cholecalciferol 400 IU/day, and intravenous calcium gluconate 2 g/day was administered on the first 2 days (Fig. 2). After 5 days of intensive calcium replacement, the total calcium concentration increased from 3.7 to 6.1mg/dL and ionized calcium from 0.591 to 0.749mmol/L, and his tremor improved. At the outpatient clinic 1 month after discharge, the total and ionized calcium levels were measured as 7.9mg/dL and 0.945mmol/L, respectively, with continuous administration of calcitriol 0.25mg/day and calcium citrate 750mg/cholecalciferol 400 IU/day.
Discussion
We report the case of a patient with TSC and severe hypocalcemia. TSC is caused by the mutation of TSC1 or TSC2 and occurs either in a sporadic or inherited form8). Hamartomatous lesions occur in multiple organs, and in relation to calcium metabolism, parathyroid adenoma resulting in hypercalcemia has been reported as a rare presentation of TSC9). Hypocalcemia in patients with TSC is also sporadically reported1011), and the cause is mainly explained by anticonvulsant drugs. Central nervous system involvement is the most common manifestation of TSC, including epilepsy (90%), subependymal nodule (80–90%), and neuropsychiatric disorders (90%)12). In particular, epilepsy is a frequent and significant cause of morbidity from infancy; thus, most patients with TSC are likely to be exposed to antiepileptic drugs for a relatively long duration.
Previous cases of hypocalcemia in patients with TSC commonly showed severely decreased calcium and 25-(OH) vitamin D3 concentration and history of anticonvulsant administration such as phenytoin1011). Suggested mechanisms of anticonvulsant-associated hypocalcemia included increased degradation of 25-(OH) vitamin D3 and 1,25-(OH)2 vitamin D3 by upregulated 24-hydroxylase CYP24A, direct inhibitory effects on calcium absorption in the gastrointestinal tract, end-organ resistance to PTH, and inhibition of calcitonin secretion713). In addition to anticonvulsant drugs, various medications such as anti-tuberculosis drugs, chemotherapeutic agents, antiretroviral drugs, and herbal medicines can affect vitamin D catabolism by the activation of the pregnane-X-receptor13). Therefore, these medications must be included in the differential diagnosis of hypocalcemia.
Our patient's laboratory findings showed substantial vitamin D deficiency with severe hypocalcemia, similar to the findings of previous studies1011). The patient had no hypomagnesemia, signs of rhabdomyolysis, or history of drugs such as bisphosphonates that inhibit bone resorption. The patient's response to treatment with calcitriol and calcium supplements was good. Although he was not taking anticonvulsants upon admission, prior prolonged use of anticonvulsants may have led to chronic vitamin D deficiency14). However, taking into account his severe hypocalcemia, his PTH level was inappropriately normal15). These findings are different from other reports and suggest the need to differentiate hypoparathyroidism from other causes. The patient had no history of prior neck surgery, and no evidence of other manifestations related to inherited hypoparathyroid disorders or autoimmune diseases. FECa is usually increased in patients with hypothyroidism due to the decreased PTH level16), which was not observed in this case (FECa<0.02). Nevertheless, the possibility of combined hypothyroidism remains due to the severely decreased serum calcium level17). The recently aggravated renal function could have contributed to hypocalcemia with hyperphosphatemia, but the presence of severe hypocalcemia could not be fully attributed to CKD progression alone18). In addition, the unidentified herbal medicine, poor nutritional status, and reduced sunlight exposure could act as causal factors. On the other hand, altered calcium metabolism is detrimental to bone health; long-term therapy with anticonvulsants can result in osteomalacia and musculoskeletal disability7). Our patient showed no noticeable bone diseases, but regular check-ups with bone mineral density evaluations and co-administration of vitamin D are required in patients receiving long-term anticonvulsant therapy.
Patients with TSC often visit the nephrology clinic with impaired renal function, and hypocalcemia is a serious medical condition in these patients as a predisposing factor for seizures. Therefore, careful workup for hypocalcemia including history of associated medication use, measurement of vitamin D, PTH, and serum concentration of currently used anticonvulsants should be performed in addition to renal care. Vitamin D administration is critical in highly at-risk patients, and bone diseases should also be evaluated.

 XML Download
XML Download