

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Fibrosis is often accompanied by repair and/or remodelling of tissues, activation of fibroblasts, formation of myofibroblasts and increase in the volume of extracellular matrix (ECM)12. Progressive interstitial and perivascular fibrosis is observed in systemic sclerosis (scleroderma, SSc)3. SSc is classified into limited SSc (lcSSc) and diffuse SSc (dcSSc) forms. Both types differ in the extent of cutaneous involvement, in which fibroblasts produce significant amounts of collagen (type I, III, VI, and VII)4. In lcSSc fibrosis is restricted to skin on fingers, digital extremities and face. Whereas in dcSSc are also affected truncal and peripheral skin areas, as well as visceral organs including interstitial lung disease (ILD), scleroderma renal crisis (SRC), gastrointestinal disease and myocardial involvement5.
NOTCH PATHWAY
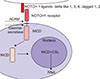
In mammals, the NOTCH pathway is conservative and consists of four receptors (NOTCH 1∼4) and five ligands: delta-like 1, 3, and 4 (DLL-1, 3, 4) and Jagged 1 and 2 (JAG-1, 2)10. The proteins DLL and JAG can effect on NOTCH receptors by cis-(synthesis of the ligand and receptor occurs in the same cell) or trans-interaction (the ligand and receptor are synthesized by different cells)11. Post-translational modifications may have a major impact on the function of NOTCH pathway proteins. Biochemical processes, like glycosylation and/or phosphorylation, may determine the specificity of a receptor to a ligand, and vice versa12. Post-translational modifications of NOTCH receptors occur in the endoplasmic reticulum. Appropriately modified receptor proteins are cut in the Golgi apparatus and transported in the form of heterodimer to the cell membrane13. Each receptor consists of three domains: extracellular, transmembrane and intracellular1214. The ligand binding domain involves the epidermal growth factor (EGF)-like repeats, which are present in the extracellular domain1415.
Once attached to the ligand, the NOTCH receptors are subject to cleavage catalysed by the metalloproteinase A Digestirn and Metalloproteinase 17 and γ-secretase. This causes the disconnection of the NOTCH intracellular domain (NICD), which is transferred into the cell nucleus, joins with the transcription factor-CSL (C-repeat/DRE binding factor 1 [CBF1]/suppressor of hairless/Lag1) and inhibits or stimulates the expression of target genes (Fig. 1)713. The CSL protein is a suppressor of transcription, while its connection to NICD results in the activation of RNA synthesis16. Low or no NICD expression is observed in normal skin fibroblasts. Its constitutive expression was described in epithelial cells: epidermal keratinocytes, hair follicle cells and sebaceous glands17181920.
SYSTEMIC SCLEROSIS
The activation of the NOTCH pathway is observed under physiological conditions, like organogenesis, and has been described in the pathogenesis of diseases associated with abnormal fibrosis, including the development of idiopathic pulmonary fibrosis, kidney fibrosis and SSc22232425. NOTCH pathway has an effect on cell differentiation, proliferation, survival and apoptosis22.
Abnormal microcirculation, fibrosis, as well as, autoimmune inflammatory processes are observed in SSc2627. Perivascular inflammation reduces vascular density, and later the ECM accumulates2728. A positive correlation between the number of T lymphocytes and the NOTCH1 gene expression in endothelial fibroblasts was found29. The differentiation of T-helper cells (Th) is affected by the NOTCH pathway ligands, including JAG-1 and JAG-2, which stimulate the immune response with Th2 cells, while DLL proteins, predominantly DLL-4, are responsible for the activation of Th1 cells4. It is assumed, that the direct interaction between T-cells expressing JAG-1 and fibroblasts showing high expression of the NOTCH1 receptor may be the main mechanism of the NOTCH pathway activation in SSc8. For example overexpression of JAG-1, Notch ligand, is observed is SSc skin and in hypertrophic scars. It induces an accumulation of NICD in SSc fibroblasts following higher expression of genes whose products are involved in fibroblast activation and collagen release24.
MYOFIBROBLAST FORMATION
The phenotype of myofibroblasts is typical for fibroblasts and smooth muscle cells30. Myofibroblasts show increased expression of collagen type I and III collagen, α-smooth muscle actin (α-SMA) and lower expression of genes encode ECM-degrading enzymes3132.
Myofibroblasts synthesize the largest amounts of ECM in tissues, where the repair or remodelling process takes place33. If the repair is completed and these cells do not enter the apoptosis, then the remaining myofibroblasts may cause scarring and fibrosis development33.
The myofibroblasts can originate from various cells, including bone marrow fibrocytes, pericytes, perivascular fibroblasts, white adipocytes, vascular endothelial cells, cholangiocytes and hepatocytes293435363738. Moreover, Dees et al.24 demonstrated that the stimulation of normal fibroblasts with recombinant human Jag-1-Fc chimera leads to a change in their phenotype, increased collagen release, differentiation of resting fibroblasts into myofibroblasts and further for development of fibrosis.
EPITHELIAL-MESENCHYMAL TRANSITION
The differentiation of epithelial cells into myofibroblasts is called epithelial-mesenchymal transition (EMT). It is a process in which epithelial cells lose their properties and show alterations in morphology, cellular architecture, adhesion and migration capacity3940. EMT occurs during organ development and in pathological conditions, e.g., in tumours or other diseases originating from fibroblasts37. EMT in kidney epithelial and intercalated cells was also described. It is the cause of tubulo-interstitial fibrosis344142. Moreover Manetti et al.43 found that EMT may take a place in the skin of patients with SSc and may have therefore a role in the pathogenesis of dermal fibrosis. They characterized the phenotype of dermal microvascular endothelial cells, which was associated with reduction in the expression of epithelial markers CD31 and vascular endothelial cadherin and an upregulation of mesenchymal markers, including α-SMA+ stress fibers, fibroblast specific protein-1 (FSP1/S100A4), type I collagen and Snail1 protein43.
Activation of the NOTCH pathway in the endothelium leads to EMT and to morphological, phenotypic and functional changes in epithelial cells44. During EMT is observed loss of expression of some genes, whose products are involved in regulation of cellular adhesion (claudins and E-cadherin) and inhibition of cytoskeletal proteins (for example SMAD6/7) to promote the mesenchymal phenotype30. Markers typical for EMT include increased expression of N-cadherin, vimentin, FSP1 and α-SMA, nuclear localization of β-catenin, decreased expression of E-cadherins and CD31 molecules39. Cytoskeletal changes promote increased cytosol mobility and the acquisition of a phenotype typical for myofibroblasts42.
Many signalling pathways, are involved in the activation of fibroblasts and EMT including transforming growth factor β1 (TGF-β1), bone morphogenic protein, EGF, fibroblast growth factor, platelet-derived growth factor, Wnt, Sonic Hedgehog and integrin signalling645464748495051. These signalling pathways activate, trough intracellular kinases, transcription factors that activate the expression of EMT-associated genes52.
Several transcription factors can be involved in the induction of EMT, among them, are the zinc-finger binding proteins Snail1 and Snail2 (also known as Slug)53. Notch signalling can regulate expression of Snail1 in a non-direcive way through induction of hypoxia-inducible factor 1α, which binds to promoter of lysyl oxidase gene following transcription of Snail1 gene54. Snail2 interacts with NOTCH receptors and is essential for Notch-mediated inhibition of E-cadherin and β-catenin genes expression55.
In addition to EMT mediated by many signalling pathways, NOTCH regulates indirectly EMT through nuclear factor kappa B, β-catenin pathways, as well as, through the action of various microRNAs56. For example JAG-2, NOTCH ligand, promotes EMT through the expression of GATA3 gene (GATA-binding protein 3), which encodes transcription factor inhibiting the cluster of microRNA-20056. Activation of the NOTCH pathway by JAG-1 in epithelial cells of breast cancer induces EMT, which leads to promoting the invasion and dissemination of malignant cells57.
INHIBITORS OF NOTCH AND NOTCH-ASSOCIATED PATHWAYS
Ligands endocytosis of the NOTCH pathway has the inhibitory effect on signal transduction. In mammals, four E3 ubiquitin ligases are involved in the process of endocytosis: neuralized-1 (Neur1), neuralized-2 (Neur2), mind bomb-1 (Mib1) and mind bomb-2 (Mib2)58. The Mib1 enzyme plays the essential role in inhibiting the NOTCH pathway, while the other ligases play an auxiliary role59. Taking into above, Choi et al.25 demonstrated that deletion of gene encoding Mib1 in kidney intercalated cells leads to increased expression of TGF-β1 and ECM volume. TGF-β1 may intensify fibrosis, for example in patients with SSc, it increases the expression of NICD and the hes family basic Helix-Loop-Helix transcription factor 1 (HES-1) protein in dermal fibroblasts (Fig. 1)246061.
Inhibition of the NOTCH pathway can be carried out by suppressing the activity of γ-secretase and blocking or reduction of the synthesis of ligands (DLL and JAG)61. The inhibitor of γ-secretase activity–DAPT (N-[N-(3,5-difluorophenacetyl)- L-alanyl]-S-phenylglycine t-butyl ester) can reduce the intensity of fibrosis62. DAPT reduces the expression of fibrosis-promoting cytokines interleukin (IL)-4, IL-6, TGF-β1 and connective tissue growth factor; and reduces the number of myofibroblasts in mice61. Moreover, Chen et al.63 demonstrated that DAPT reduced the number of cells with characteristics of myofibroblasts and inhibited the expression of TGF-β1. In SSc, blocking the NOTCH pathway by applying DAPT prevents the development of skin fibrosis (bleomycin-induced) in in vitro models dependent on inflammation (dermal fibrosis in inflammation-dependent models) or in animal models, e.g., the tight skin 1 (Tsk-1) mice2461. Targeting of NOTCH pathway trough anti-sense RNAs or by DAPT can reduce the collagen release in SSc fibroblasts, without any negative effects on wild type fibroblasts24. Furthermore, inhibition of Notch signalling prevent the development of fibrosis in different models, among them, bleomycin-induced fibrosis and in the Tsk-1 mause model761.
In SSc skin is observed JAG-1 overexpression, which increased expression of targeted genes in various tissues16. Inhibition of the NOTCH pathway by knockdown of JAG-1 leads to the inhibition of keloid fibroblast proliferation and migration, and has antiangiogenic activity64. The direct effect on the NOTCH pathway results in the reduction of collagen synthesis by fibroblasts and may be effective both in the early proinflammatory stages of SSc and in later phases not related to the inflammatory process2461.
LIVER FIBROSIS
The mechanism of tissue fibrosis has been well understood in the liver. Its fibrosis occurs as a result of chronic liver disease. In SSc is observed excessive fibrosis in the viscera and activation of hepatic stellate cells (HSCs) after liver injury65. Activated HSCs are characterised by increased expression of α-SMA and can be transformed into myofibroblasts66. Activation/auto-activation of HSCs increases fibrosis and biosynthesis of JAG-1 ligand and the expression of the Notch3 gene63. Unactivated HSCs do not show an expression of both JAG-1/2 ligands67. All four types of NOTCH receptors are expressed on the surface of healthy liver cells. Liver fibrosis increases expression of NOTCH3 protein/Notch3 gene6869. The expression of Notch1, Notch2 and Notch4 genes is at a similar level in both healthy and fibrotic liver cells69.
KIDNEY FIBROSIS
Kidney involvement is SSc is primarily manifested by SRC. It is defined as the accelerated arterial hypertension and/or rapidly progressive oliguric renal failure. The primary process, associated with SRC, is injury to the endothelial cells70. These cells are found in vascular, glomerular and peritubular capillary (PTC) beds71. Kidney endothelial cells, which underwent EMT, play an important role in the fibrosis of this organ72.
Kidney fibrosis is the hallmark of chronic kidney disease (CKD). It is characterized by the accumulation of myofibroblasts and excessive deposition of ECM components and the tubulo-interstitial fibrosis257374. The kidney fibrosis process is accompanied by the process of repairing PTCs. However, renal PTCs are very susceptible to atrophy occurring after this organ has been damaged75. The concomitance of PTCs with interstitial fibrosis is one of the main symptoms of CKD. Animal models showed a negative correlation between the density of PTCs and the intensity of fibrosis75. In patients with CKD, the increased loss of PTCs is a factor associated with increased interstitial fibrosis and kidney failure4276. The repression of Notch1 signalling counteracts PTC rarefaction75.
LUNG FIBROSIS
ILD is observed in up to 50% of SSc patients2881. Pulmonary fibrosis is associated with dysfuntion of epithelial cells, accumulation of fibroblasts, increased production of TGF-β1, excessive deposition in ECM and abnormal lung remodeling8283. Idiopathic pulmonary disease is characterized by myofibroblast formation (by EMT), which is facilitated by activation of NOTCH pathway84. Liu et al.9 indicated, that TGF-β1 stimulated expression of JAG-1, Notch1, NICD and HES-1 following the differentiation of rat primary lung fibroblasts.
HEART FIBROSIS
NOTCH pathway is a key mechanism of normal heart morphogenesis. It regulates cardiomyocyte proliferation, formation of valves, atrioventricular canal, outflow tract and coronary vessels8586.
Cardiac fibrosis is characterized by excessive deposition of scar tissue Cross-talk between NOTCH and TGF-β pathways play an important role during the development of the heart. Furthermore, NOTCH pathway is involved in the heart fibrosis after myocardial infarction (MI), primarily through myofibroblast differentiation87.
NOTCH receptors and their ligands are localized to the vasculature, as well as, are observed in endocardium. JAG-1, which is NOTCH ligand, is present on endocardial and periendocardial cells of the cardiac cushions888990. In vivo study of Boopathy et al.91, showed that delivery of JAG-1 ligand through intramyocardial injection in rats with MI reduced cardiac fibrosis. The inhibition of NOTCH receptors expression-1, 3, and 4 promotes fibroblast- myofibroblast transition87. Moreover, Zhang et al.92 found in animal model that overexpression of Notch3 receptor (as a result of cDNA lentivirus inections) increased mice survival rate, improved cardiac function and minimized MI-induced increase in cardiac fibrosis.
SUMMARY
The ever growing evidence suggests that the NOTCH pathway is involved in the development of fibrosis in various organs761. However, the molecular mechanisms associated with this process have not been fully recognised. It is possible that inhibitors of NOTCH receptors or their ligands will be used in the future in new therapeutic strategies in SSc patients. Furthermore, there is insufficient data on the expression of NOTCH receptors, the occurrence of polymorphisms and possible mutations in genes which encode the above receptors in fibrotic tissues. The research in this area is still to be continued.
 XML Download
XML Download