

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Dear Editor,
Spinal muscular atrophy (SMA) is caused by homozygous deletion of the survival motor neuron gene (SMN1), and copy-number variation (CNV) in the SMN2 gene is thought to influence the disease severity.1 We present two late-onset SMA siblings who manifested with marked differences in clinical severity and muscle imaging despite the same copy numbers of SMN2.
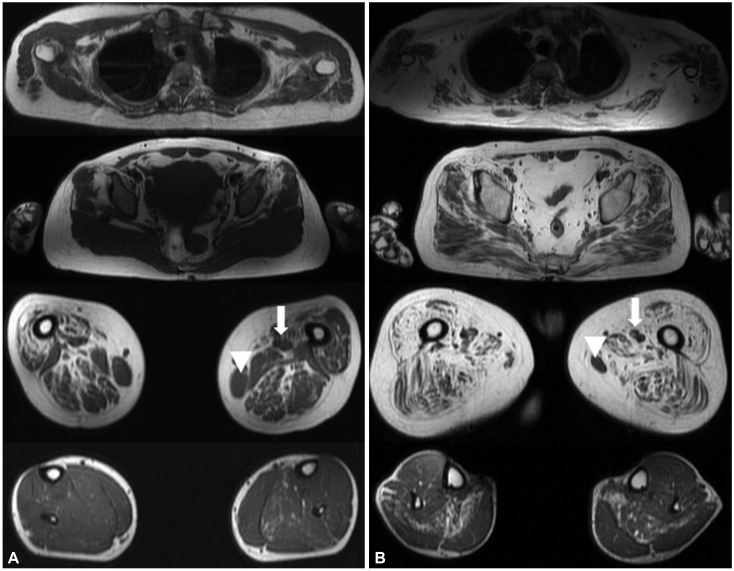
A 43-year-old female presented with proximal limb weakness since high school that had not deteriorated until she reached 40 years of age. The initial neurological examination of the proband demonstrated proximal limb weakness in the lower extremities, at MRC grade 4. However, she had no difficulty in performing the activities of daily living, but experienced difficulties during strenuous exercise such as hiking. The proband's elder brother was 45 years old, and he had found it difficult to run during high school, and had been incapable of independent walking since his 30s. The initial neurological examination revealed a proximal limb weakness with the lower extremity being affected more, and he was nonambulatory. Muscle MRI revealed that the proximal thigh muscles were markedly affected with relative sparing of the adductor and gracilis muscles (Fig. 1A). Her muscle biopsy showed fiber-type grouping, implicating neurogenic changes (Supplementary Fig. 1 in the online-only Data Supplement). The muscle MRI of the proband's elder brother illustrated a diffuse fatty change in proximal muscles including gluteus maximus, gluteus medius vastus, and hamstring muscles, but the gracilis and adductor longus muscles were mildly spared (Fig. 1B). A gene study of SMN1 revealed deletion of exons 7 and 8, confirming the diagnosis of SMA; the proband was classified as SMA4, while it was more appropriate to classify her brother as SMA3b. We measured the copy number of SMN2 of these two siblings using two different methods to ensure a precise diagnosis. We compared the SMN2, NAIP, and CFTR copy numbers as described previously,1 and we additionally used droplet digital PCR analysis (Bio-Rad Laboratories; ddPCR SMN2 copy number determination kit, Hercules, CA, USA). The obtained results were further confirmed by multicopy marker analysis as described by Melki et al.2 Both patients showed four copies of SMN2 and the same NAIP and CFTR copy numbers. Next-generation sequencing was also performed to screen the variants in DYNC1H1, BICD2, SMN1, PLS3, and NCALD, but this revealed no pathogenic mutation or known modifier known to affect the phenotype of our patients.
The CNVs of SMN2 are generally known to be correlated with the phenotype.1345 However, there are reports of phenotype discrepancies in patients with same copy numbers of SMN2.45 This means that variability in the phenotype cannot be reliably explained only by CNVs, and so we expect that there are other unknown genetic modifiers, sex differences, or epigenetic factors. It has reported that there are sex-related differences in severity or in intrafamilial phenotype variability.4678 A recent clinical study of myotonic dystrophy type 1 showed that sex differences might be another modifying factor influencing the clinical profile and severity of the disease.6 Consistent with these studies, we observed a significantly more severe phenotype in the male patient but a mild phenotype in the female patient of the same family. Therefore, even in SMA, sex difference is a possible modifying factor associated with phenotype variability, and further studies are warranted to clarify this.
 XML Download
XML Download