

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
GNE myopathy is a muscle disease caused by biallelic recessive mutations in the gene encoding UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase (GNE).1 It encompasses all of the previously described terms of distal myopathy with rimmed vacuoles, Nonaka myopathy, hereditary inclusion body myopathy, and inclusion body myopathy type.2 The term GNE myopathy was proposed as being more appropriate for unifying all of the above myopathic conditions related to GNE mutations.2
GNE myopathy starts during adulthood, most frequently in the third decade, and initially involves distal leg muscles, with slow progression to the proximal legs and upper limbs.2 Cardiac and respiratory functions are typically spared. Some cases present with proximal leg weakness,3,4 finger weakness,5 or calf atrophy.4 Rimmed vacuoles on muscle pathology are a clue for a diagnosis of GNE myopathy, while it is not detected in all cases of genetically confirmed GNE myopathy. We have previously reported that a significant number of patients present with limb-girdle weakness, with some patients not having rimmed vacuoles in muscle biopsies.3 The quadriceps muscles are usually spared even in the advanced stages, frequently exhibiting normal or minimal histopathology.
More than 150 pathogenic GNE mutations have been detected, most of which are missense mutations dispersed over the entire length of the GNE gene.6 Some mutations appear at higher frequency in certain ethnic groups, including p.Met743 Thr in Iranian Jewish patients and p.Val603Leu in Japanese patients.7
Supplementing intermediate metabolites of the sialic acid pathway including sialic acid, sialyllactose, and ManNAc has shown promising results in preclinical studies.8 Unfortunately, the extended-release form of sialic acid failed to show effectiveness in the phase 3 clinical trial (clinicaltrials.gov: NCT 02377921), indicating a gap in the translation of the success in mice to humans. Clinical trial using ManNAc as an oral agent are currently underway.9
Clinical profiling and determining the natural history are more important than ever for maximizing the confidence in a clinical trial. In the present study we analyzed the clinical and genetic profiles of Korean patients with genetically confirmed GNE myopathy. We also investigated the correlations between clinical features, mutations, and disease severity.
Go to : 
METHODS
Patients
This study included 44 patients (19 males and 25 females) with homozygous or compound heterozygous GNE mutations, most of which were previously reported as pathogenic ones and others were predicted as being ‘damaging’ by in silico analyses, and also classified as ‘pathogenic’ or ‘likely pathogenic’ according to the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP) guideline.10 All of the recruited patients agreed to provide their medical records, complete clinical questionnaires, and provide written informed consent. The Institutional Review Board of Pusan National University Hospital approved this study (IRB No. H-1712-002-061).
GNE mutation data
All of the included patients had confirmed pathogenic GNE mutations in nine different hospitals with neuromuscular specialists prior to recruitment to this study. We extracted DNA from the whole blood of 15 of the patients who directly visited our neuromuscular center, which was used in PCR amplification and direct sequence analyses in our laboratory. The PCR amplification covered all 12 exons and the immediately flanking intronic regions of the GNE gene. The PCR products were sequenced in both the forward and reverse directions. Sequences were analyzed and compared with the reference sequence (GenBank accession number NM_001128227.2). Seven of the patients who were genetically diagnosed in other hospitals personally provided their medical records, including mutation data. For the remaining 22 patients who could not acquire their mutational reports from their hospitals, GNE tests were performed at the laboratory of EGL Genetics (Tucker, GA, USA).
Functional status
We applied a questionnaire-based survey to determine the functional status of the patients. Most of the information was collected using a self-reporting questionnaire, but we also retrospectively reviewed any available hospital records for further information. The questionnaire included inquiries on the following features: age at disease onset, initial presentation symptoms, mode of ambulation, and time of losing ambulation. In addition, we asked how many of the following eight major joint movements of the body could be performed against gravity: shoulder abduction, elbow flexion, wrist flexion/extension, hip flexion, knee extension, ankle dorsiflexion, neck flexion when supine, and trunk flexion when supine. Patients were asked whether they were independent or dependent on caregivers when performing major activities of daily living, including standing from a squatting position, getting up from the bed, eating, cleaning the face, going to the toilet, and changing clothes.
Statistical analyses
Linear regression analysis was applied to the relationships between disease duration and joints with mobility against gravity, and between disease duration and independent daily activities. Subjects capable of either all or none of the functional movements and daily activities were excluded from the analysis in order to prevent the ceiling effect. We further evaluated the disease progression depending on the initial symptoms or the domains of GNE mutations. These subgroup analyses were performed using Student's t-test or one-way ANOVA. A p value of <0.05 was considered to be indicative of statistically significant data.
Go to :
RESULTS
General characteristics
The clinical information of the 44 Korean patients from 41 families is provided in Table 1. Their age at the time of data collection was 36.0±10.7 years (mean±SD). Their age at onset ranged from 14 to 51 years (26.3±9.2 years), and patient 16-II was an asymptomatic brother of patient 16-I. The disease duration from onset to diagnosis was 1–41 years (10.0±8.8 years). Eleven of the patients had affected siblings compatible with an autosomal recessive inheritance pattern. None of the patients with a detailed family history reported obvious consanguinity.
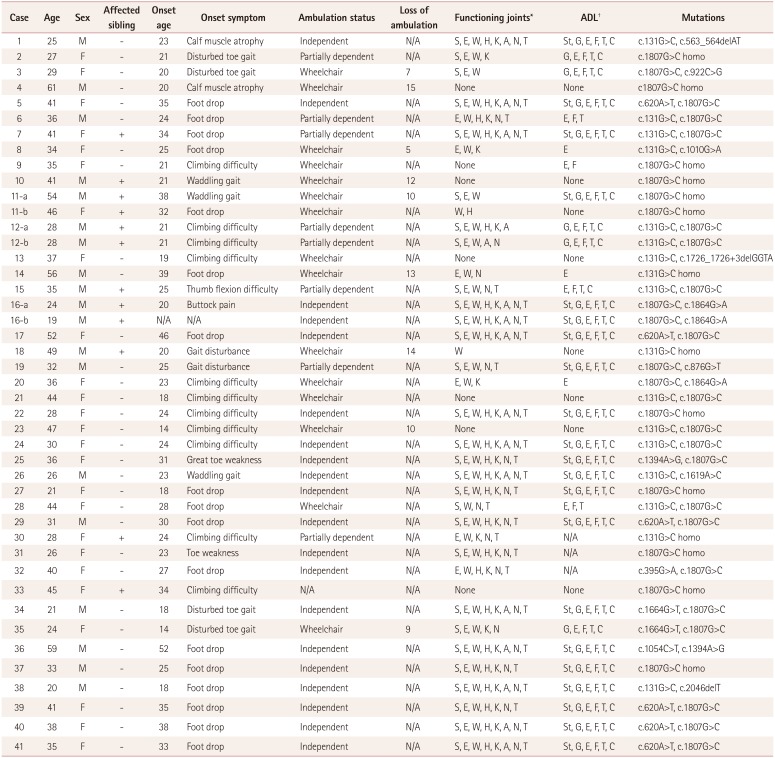
Table 1
Clinical profiles of all subjects in this study
![]()
GNE mutations
All of the recruited patients harbored homozygous or compound heterozygous GNE mutations. The types of mutations and their locations and frequencies are presented in Table 1 and Fig. 1. The most common mutation was c.1807G>C (p.Val603Leu), which was found in 45 alleles (51.1%), followed by c.131G>C (p.Cys44Ser) found in 20 alleles (22.7%). Homozygosity was also related to these two common mutations, and was found in 11 patients with p.Val603Leu and 3 with p.Cys44Ser. Two of the 15 different mutations have not been reported elsewhere. One of the novel mutations (c.1619A> C, p.His540Pro) was predicted to be ‘damaging’ and ‘disease causing’ by in silico analyses implemented using PROVEAN (http://provean.jcvi.org) and MutationTaster (http://mutationtaster.org), and classified as ‘likely pathogenic’ according to the ACMG/AMP guideline, since a change in the same amino acid by a different nucleotide change (c.1618C>T, p.His540Tyr) is reportedly pathogenic.11 The second one (c.2046delT, p.Val 684Leufs*2) can be classified as ‘pathogenic’ since it is expected to cause premature termination by frameshift. The other 13 mutations were first reported in Korean, Japanese, or Chinese patients.6 Except for 3 frameshift mutations involving small deletions, 12 were all missense mutations. No case had truncating mutations in both alleles. The mutations were scattered throughout the exons. Eight of the mutations were located in the epimerase domain (ED) and the other seven were in the kinase domain (KD), as presented in Fig. 1. Both mutations were present in KD in 17 of the patients (38.6%), while both were in ED in 5 patients (11.4%). The other 22 patients (50.0%) had one mutation in each domain (ED/KD).

Presenting symptoms; initially affected muscle groups
Various initial symptoms at disease onset were reported by the patients, as indicated in Table 1. Foot drop and/or big-toe extension weakness indicating affected anterior lower leg muscles was reported in 19 of the patients (43.2%). Thirteen patients (29.5%) reported running or climbing difficulty suggestive of hip-girdle weakness as their first symptom. Six of the patients (13.6%) described difficulty in toe gait or calf atrophy corresponding to the posterior lower legs being affected. One exceptional patient reported weak thumb flexion as his first symptom. Patient 16-II remained asymptomatic until the last follow-up at an age of 19 years. The remaining four patients recalled experiencing nonspecific symptoms (e.g., buttock pain or gait disturbance) at the beginning of their disease.
Among 20 patients for whom medical records on the initial symptoms were available, 12 described having more prominent weakness in ankle dorsiflexion. Meanwhile, disturbed toe gait and/or calf muscle atrophy was reported in two of the patients, while proximal dominant leg weakness was described in the other four.
Decrease in the number of functional joints
We considered a set of joints to be functional when the patient could flex or extend the joint against gravity. Fig. 2A depicts the pattern of loss of functional joint movement. Ankle dorsiflexion was the most commonly affected movement when the patient had just begun to lose a single functional set of joints. On the other hand, wrist or neck flexion was most likely to be the last functional movement that was lost.

All 8 sets of joint movements remained functional in 12 of the patients, and their disease duration was 2–7 years (4.2±2.2 years). Seven patients had no functionality in any of the eight considered joint movements, and their disease duration was 11–41 years (23.3±10.7 years). The number of functional joint sets was strongly correlated with the disease duration, as shown by the results of the linear regression analysis in Fig. 2B (r2=0.5228, p<0.0001) reflecting the progression of GNE myopathy. The mean yearly rate of decrease in the number of functional joint sets was −0.2581, with a 95% CI of −0.1517 to −0.3645.
Ambulation
Sixteen patients (36.4%) had to use wheelchairs throughout the day, 20 patients (45.5%) were still capable of completely independent ambulation, and 8 patients (18.1%) used walking aids when walking longer distances. The self-reported disease duration to a wheelchair-bound state was 10.6±3.3 years (5–15 years). There was a clear difference in the disease duration between ambulatory and wheelchair-bound patients, at 4.5±0.7 years (1–13 years) and 17.4±2.2 years (9–41 years), respectively (Fig. 3A).
 | Fig. 3Functional status during ambulation and daily activities. A: The disease duration clearly differed between the ambulatory and wheelchair-bound patients, at 4.5±0.7 and 17.4±2.2 years, respectively. B: All of the daily activities that could be performed independently by each of the patients. C: The number of independent daily activities was correlated with the disease duration (p=0.012). ADL: activities of daily living.
|
Functional status during daily activities
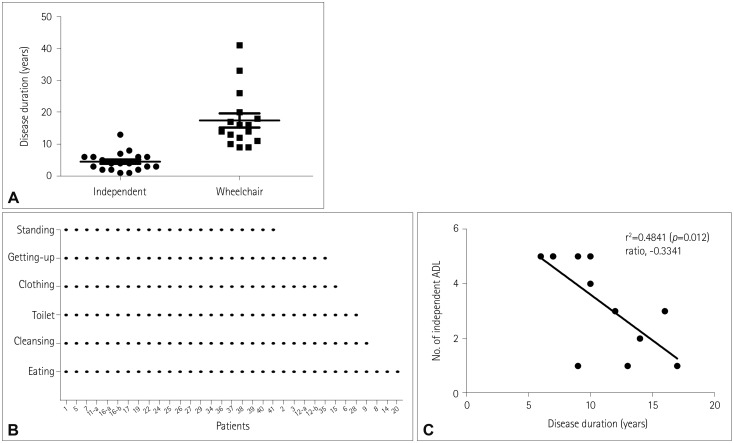
The daily activities that each patient could perform independently are listed in Fig. 3B, sorted in decreasing numerical order. Among 41 patients who provided information on their independent daily activities, 21 (51.2%) were could still independently perform all six kinds of the activities surveyed. Standing from a squatting position was disturbed at the earliest time point, which was lost in all five patients who could still perform the other five activities. The disease duration of these patients was 6–10 years (7.8±1.6 years).
After standing from a squatting position, the order of independent activities lost were getting up from the bed, changing clothes, and going to the toilet, all of which require movements of both the upper and lower extremities. Cleaning the face and eating, which mainly involve the upper extremities, were the last activities to be disturbed. Eight patients (19.5%) could perform none of the activities at all, and they had a disease duration of 11–41 years (24.0±10.1 years). The relationship between disease duration and independent activities is depicted in Fig. 3C (r2=0.4841, p=0.012).
Disease severity according to types of initial symptoms
The age at onset was markedly younger in the subgroup with limb-girdle phenotype (23.2±1.6 years) than in that with foot drop onset (31.2±2.0 years, p=0.006; Fig. 4A). The duration from the onset to the diagnosis was significantly longer in the subgroup with limb-girdle (5.1±0.9 vs. 11.1±2.6 years, p=0.0186), as shown in Fig. 4B.
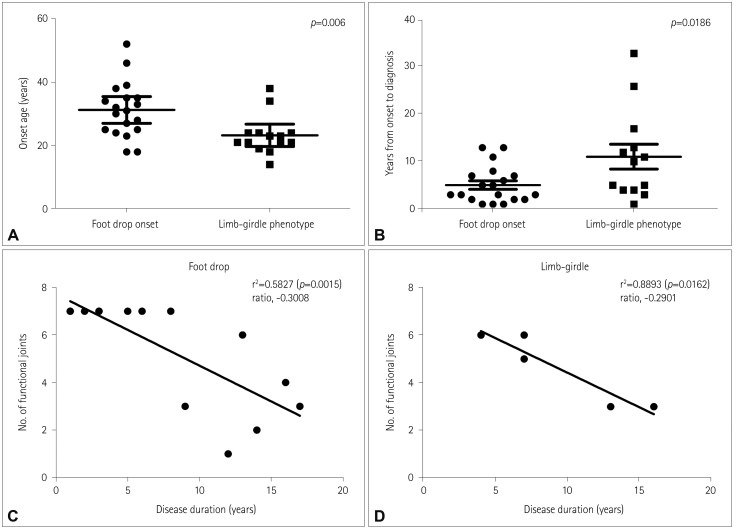 | Fig. 4Disease severity according to the types of initial symptoms. A: The onset age was significantly younger in the subgroup with limb-girdle phenotype. B: The duration from the onset to the diagnosis was significantly longer in the subgroup with limb-girdle phenotype. C and D: Disease progression as represented by the functional joint sets according to disease duration did not differ between the subgroups of foot-drop onset and limb-girdle phenotype (p=0.5731).
|
The yearly rates of loss of functional joints were −0.3008 (r2=0.5827, p=0.0015) and −0.2901 (r2=0.8893, p=0.0162) in the subgroups of foot drop and limb-girdle respectively, with no significant difference (p=0.5731) (Fig. 4C and D).
Disease severity according to domains of GNE mutations
We compared the disease severity among subgroups according to domains where the mutations were located; that is, the KD, ED/KD, and ED subgroups. The age at onset did not differ significantly among these subgroups (p=0.387) (Fig. 5A). On the other hand, the decrease ratios of functional joints did differ significantly among the three subgroups (p=0.0062, Fig. 5B–D), with a mean decrease ratio of -0.3617 in KD (r2=0.7234, p=0.0037), −0.2601 in ED/KD (r2=0.4403, p=0.0186), and −0.1402 in ED (r2=0.8760, p=0.0641). The disease duration among wheelchair-bound patients did not differ significantly between the two subgroups of KD (17.5±3.6 years, n=8) and ED/KD (15.9±3.4 years, n=8; p=0.7455). The ED subgroup was excluded from this analysis due to the small number of wheelchair-dependent subjects (n=3).
 | Fig. 5Disease severity according to the domains of GNE mutations. A: The onset age did not differ significantly among the three subgroups of KD, ED/KD, and ED. B–D: Disease progression as represented by the number functional joint sets according to disease duration significantly rapid in the order of KD (B), ED/KD (C), and ED (D) (p=0.0062). ED: epimerase domain, KD: kinase domain.
|
Go to :
DISCUSSION
This study included 44 Korean patients with GNE myopathy who agreed to submit their genetic report biallelic GNE mutations. The missense mutation p.Val603Leu was the most common pathogenic allele (51.1%), followed by p.Cys44Ser (22.7%). The most frequent genotypes were homozygous p.Val603Leu (n=11) and the compound heterozygotes of p.Val603Leu and p.Cys44Ser (n=9). These findings are consistent with those of previous studies of Korean patients with GNE myopathy.3,12,13 Although known GNE mutations are scattered throughout the gene, these two variants comprise more than half of all cases. These mutations also appear very frequently in Japanese patients, corresponding to the first and third most common mutations.14 Four of the mutations in the present study have been reported only in Korean patients with GNE myopathy,3,13 and the remaining ones are shared with Japanese and/or Chinese cases.6 However, the mutation of p.Asp207Val, which is the second most common mutation in Japanese patients, was detected among a smaller proportion (6.8%) of our cohort, and p. Val603Leu has been found in only a few Chinese patients.15 The homozygosity found in 14 patients either with p.Val603Leu and p.Cys44Ser was not related to consanguinity according to the best knowledge of the patients; marriage to a third cousin or closer relative is prohibited both culturally and legally in Korea. The allele frequencies of the two most common variants estimated by homozygosity are 0.046% and 0.024%. The allele frequency of the p.Val603Leu variant reported by the Exome Aggregation Consortium is 0.034% (3/8, 654) in East Asians.
Clinical profiles were analyzed with the aid of a questionnaire-based survey and complemented by reviewing the available medical records. The general findings from our questionnaire survey were comparable to those obtained from the analysis of the medical records and registry data. The onset age of our patients was similar to that in another Korean cohort (23.8±8.8 years),13 a Japanese cohort (25.2±9.2 years),16 and a recent global study involving subjects with various ethnic backgrounds (mean 27.8 years).17 In terms of age at data collection, one Japanese cohort included older patients (age 43.1±10.7 years), probably with a longer disease duration,18 and our patients (age 36.0±10.7 years) were as old as those in the recent global study (mean ages of 38.7 years in males and 39.5 years in females).17 The disease duration from onset to the time of using wheelchair was slightly shorter in our cohort (10.6±3.3 years) than in the other studies (11.5±1.2 years in Japan and a mean of 11.9 years in the global study),17,18 which may have been due to the national support program for assistance devices in Korea.
Compared to our previous report on 11 patients,3 a smaller proportion of the present patients reported proximal leg weakness as their first symptom, while it was significantly larger than previously estimated. A particularly interesting observation was that two patients in this cohort lost all functional movements in the hip, neck, and trunk prior to experiencing weakness in ankle dorsiflexion. Some patients presented with atypical patterns of weakness, such as toe-gait difficulty and finger weakness. These findings support the presence of heterogeneity in the clinical course of GNE myopathy, which clinicians should be aware of during the diagnostic process of genetic myopathies.
The patient-reported functional status as represented by the number of functional limb movements and independent daily activities showed a strong correlation with the disease progression in the present study. A recent study also found that patient-reported outcomes were valid and reliable measures of physical function in GNE myopathy.19 According to a previous report on the GNE Myopathy Functional Activity Scale (GNEM-FAS), which was developed for clinical research and trials,20 mobility, upper extremity function, and independent activities of daily living were most affected by the disease progression in GNE myopathy patients. Our questionnaire included items that were scored on the GNEM-FAS, although we did not quantify motor power in the upper or lower extremities. Counting the number of functional limb movements and the activities of daily living—as in the present study—are useful methods for assessing the functional status.
Our results reveal marked deteriorations in functional limb movements and daily activities over a disease duration of 10 to 20 years. These data will facilitate predictions of the order of increasing disabilities, and thereby guide patients and their caregivers to plan ahead. During the first decade of disease onset, ankle dorsiflexion and standing from a squatting position were the most frequently affected movements. Wrist and neck flexion as well as the activities of the upper extremities such as cleaning the face and eating were preserved up to the most advanced stages. The patients lost all of their functional joints and became fully dependent on caregivers for daily living at 30 years after the onset. Patients with limb-girdle weakness had an earlier onset and a more-delayed diagnosis compared to those with foot-drop onset. However, we did not see a significant difference in the rate of progression.
GNE is a distinctively bifunctional enzyme, and the position of GNE mutations may influence the phenotype. In our subgroup analyses, the age at onset did not differ among the KD, ED/KD, and ED subgroups. However, disease progression as represented by the decrease in the ratio of functional joints was significantly more rapid in the KD group than in the ED/KD and ED groups. One Japanese and another Chinese report also described that patients with heterozygous ED/KD mutations show a less severe phenotype,15,18 while another report on patients with various ethnic backgrounds demonstrated that the ED/KD genotype tended to be associated with the lowest proportion of ambulatory patients, earliest onset, and earliest use of a wheelchair.17 That report pointed that p.Asp207Val, which is known to be a milder mutation, was predominant in Japanese and Chinese cohorts, and possibly obscured the final analysis. However, it is unlikely that our results were affected by the mild p.Asp207Val mutation, since our cohort included far fewer patients with p. Asp207Val. Thus, the present study reinforces that the ED/KD genotype, which theoretically involves each allele compensating for the other, is related to a less severe phenotype.
In conclusion, this study has revealed the genetic and clinical characteristics in a Korean cohort of GNE myopathy, especially in relation to the disease severity and progression. This is the first large-scale report on Korean GNE myopathy, and it has provided basic data on the natural course of GNE myopathy, which will be helpful not only for managing care but also for designing future clinical trials. A limitation of this study is its retrospective analysis based on self-reporting questionnaires. Future prospective monitoring of disease severity with quantitative measurements will provide less biased insight into the natural course.
Go to :
 XML Download
XML Download