

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Dementia is an emerging global public health challenge. Currently about 50 million people, or roughly 5% of the world's older population, are affected by dementia worldwide. This number is expected to double by 2030 and more than triple by 2050.1 With these figures in mind, most countries have developed and implemented their own action plans for dementia. Recently, the World Health Organization (WHO) has published a global action plan of public health response to dementia for 2017–2025.2 The WHO's global action plan comprises the following seven action areas: 1) dementia as a public health priority, 2) dementia awareness and friendliness, 3) dementia risk reduction, 4) dementia diagnosis, treatment, and care, 5) support for dementia caregivers, 6) information systems for dementia, and 7) dementia research and innovation.2 In 2015, global dementia costs were estimated at US$ 818 billion, equivalent to 1.1% of global gross domestic product, ranging from 0.2% for low-income and middle-income countries to 1.4% for high-income countries. By 2030, it is estimated that the cost of caring for people with dementia worldwide will increase to US$ 2 trillion. This could undermine social and economic development and overwhelm health and social services, including long-term care systems.3 The number of people with dementia among elderly Koreans aged ≥65 years has been reported to be 661,707, with a prevalence of 9.8%. The management cost per dementia patient was estimated to be 205.4 million Korean won and the national dementia management cost was estimated to be 13.589 trillion Korean won, accounting for 0.8% of the gross domestic product.4
Alzheimer's disease (AD), the most common form of dementia, has been estimated to account for 60%–70% of all dementia cases. Other major forms of dementia include vascular dementia (VD), dementia with Lewy bodies, and frontotemporal dementia (FTD). Boundaries of clinical manifestations between different forms of dementia are indistinct. Mixed forms of AD and VD and other overlapped forms of neurodegenerative diseases (ND) are sometimes present.5 The concept of overlapping syndromes is now widely accepted for various ND, including AD, FTD, amyotrophic lateral sclerosis (ALS), Parkinson's disease (PD), and other degenerative diseases affecting the basal ganglia.6
With recent advances in genetic and phenotypic-pathological comparative studies in the field of ND, traditional categories of ND have been called into question. Indeed, each disease entity is no longer considered as a single disease. Instead, it is considered as complex spectral syndromes with diverse neurobiological and pathophysiological mechanisms. Because of complexities in cell death mechanisms of ND, single mechanism-targeted therapeutic strategies for treating ND such as AD, PD, and ALS have largely failed in late-stage clinical trials.
Considering diverse pathophysiologic mechanisms and various biomarkers associated with neuropathological and heterogeneous clinical phenotypes, the development of new therapeutic strategies such as stratification and personalized medicine could be the future direction of clinical trials. Therefore, platform development of new drug discovery based on novel reliable biomarkers is desperately required. These biomarkers should reflect clinical phenotype, genetic information, and neuroimaging findings.
In this review, we will describe the importance of theragnostic strategy for AD and related dementia focusing on immune-inflammatory modulation by adopting stratification and the concept of personalized medicine.
DEVELOPMENT OF THERAPEUTIC TARGETS AND DRUG DISCOVERY: CURRENT AND FUTURE DIRECTIONS
AD is a heterogeneous complex syndrome with diverse clinical manifestations, including its progression speed and clinical characteristics.7 However, most currently available medications for AD only have effects on its symptoms.8 These medications include symptomatic cognition enhancing agents such as cholinesterase inhibitors (donepezil, galantamine, and rivastigmine) and NMDA receptor antagonist (memantine). While these have recently been reported to possess additional neuroprotective effects, their efficacies are still limited.8
Traditional or current processes of drug development can be summarized as follows. A drug candidate is tested for safety and tolerability in a phase 1 clinical trial. The relationship between the dose of a drug and biological activity is then investigated in a phase 2 trial, followed by a larger phase 3 trial to assess its safety-efficacy profile. In clinical practice, healthy volunteers (phase 1) or patients (phases 2 and 3, or phase 1 for rare diseases) are monitored by health care providers in hospitals or clinical units and major outcomes are measured based on clinical findings or laboratory tests, including biomarkers (Fig. 1).9
Fig. 1
A comparison of drug development (A) and clinical trial paradigms (B) between the traditional model and future directions (modified from pharma 2020: the vision18).
CIM: confidence in mechanism, CIS: confidence in safety.
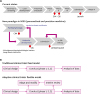
Failure of recent clinical trials can be explained by several aspects. Most clinical trials have focused on single-target drug development that is based on the classic amyloid hypothesis and/or tau-hypothesis. Furthermore, there is a lack of appropriate biological markers that are correlated with clinical outcomes and complex pathophysiological findings.10 The most important downfall of this traditional approach is the “one size fits all” principle. That is, enrollment criteria for clinical trials are largely based on clinical phenotype whereby biomarker candidates are validated against a plethora of heterogeneous clinical operationalized syndromes rather than against genetically and biologically determined entities.11 Future work should therefore aim to meet standards of Predictive, Preventive, Personalized, and Participatory (P4) medicine. Personalized medicine is one component of the P4 medicine. It aims to tailor treatment for individual patients, unlike the traditional approach.11
Therefore, optimal drug development for AD and ND should include concepts of systems biology and systems neurophysiology that allow for delineation of multivariate and combinatorial profiles of genetic, biological, pathophysiological, and clinical markers reflecting the heterogeneity of this condition. This new technology could represent a future direction of drug development for AD and ND.12
Compared to other diseases, drug discovery of AD has a high failure rate.13 The design of clinical trials can be improved based on lessons learned from past trials. Comprehensive interpretation of safety issues and selection of the most appropriate therapeutic targets from preclinical animal data are essential for drug development. However, there are some crucial factors that could lead to a better success rate in drug development for AD. These include a better understanding of the pharmacodynamics of a target drug and proof of concept from phase I and phase II trials, identification of appropriate doses and sample size, an accurate diagnosis of AD with biomarker selection, and compatible primary outcome measurements reflecting significant clinical efficacy that is directly associated with target efficacy.13
Recently, robust pharmacodynamics biomarkers of drug efficacy have been reported to be positively correlated with drug approval.14 Therefore, the Right target, Right tissue, Right safety, Right patient, and Right commercial potential (5R) framework is strongly recommended to outline key factors for successful drug discovery.14 In other words, the efficacy and pharmacokinetic/pharmacological mechanisms of therapeutic agents should be demonstrated using reliable biological markers that could reveal a strong link between the target and disease. Enrollment or stratification of the most responsive patient population is also an important factor for successful clinical trials.
Recent evidence has shown that the success rate of clinical trials increases when a stratification or enriched model is used. Selecting reliable biological markers that reflect target engagement of agents13 and using a genetics-based clinical design are examples of how hurdles of current clinical trials can be overcome.15
IMPORTANCE OF STRATIFICATION AND A FAST-TRACT STRATEGY FOR DRUG DEVELOPMENT
Patients with AD phenotype show heterogeneity in their clinical signs, biomarkers, cognitive profiles, and disease progression rates.16 This heterogeneity has allowed various categories to be established, including early-onset or late-onset, familial, and rapidly declining forms.16 Nevertheless, few comprehensive studies have characterized AD subtypes based on clinicopathologic-molecular findings including biomarkers.16
For example, in a clinical setting, approximately 10% to 30% of AD cases represent rapid progressive AD and some genetic mutant forms of FTD show a rapid progressive clinical course.11 Therefore, it is important to delineate clinical-biological characteristics of the subpopulation with rapid AD progression and disease heterogeneity. Although the definition of “rapid progression” might be differently used in previous studies, an MMSE score decrease of 6 points per year has been suggested as an indicator of rapid progression.17
Even within this selected subpopulation, patients might have different genetic and pathophysiological characteristics. Therefore, these populations should be re-grouped in a stratified manner that includes genetic profiles, imaging findings, precise clinical characteristics, and standardized biomarkers. These steps are essential in the design of future clinical trials with high success rates. Furthermore, the importance of sexual dimorphism in stratification should not be neglected in future drug development for AD.12
By 2020, we believe that all medicines that receive approval will be approved on a real-time basis, with live licenses contingent on results of extensive in-life testing, including trials with specific patient subpopulations according to a predetermined schedule for reviewing each set of results. If in-life testing confirms that a medicine is safe and effective, the company making it will be granted an extended license or special permit so that it will have an incentive to conduct further studies. In other words, every medicine on the market will have a prearranged, fully automated pathway throughout its lifecycle. Its development will be a continuous process rather than ending once it is approved (Fig. 1). This is the new paradigm in 2020. It is based on personalized and precision medicine.18
The concept of adaptive clinical trials was recently developed. Traditional clinical trials are straightforward but inflexible as they do not include options for changes of clinical protocol that may become desirable or necessary during the course of the trial. The most important advantage of adaptive designs is that clinical trials are flexible. They can add an interim review system that can adapt and modify the clinical design to linear design–conduct–analysis sequence (Fig. 1B). Adaptive designs also allow for a scheduled interim for data inspection while the trial is ongoing. Pre-specified changes to the trial's course can also be made based on analyses of these accumulating data whilst maintaining validity and integrity of the trial.19
The Korean AD Research Platform Initiative based on Immune-inflammatory biomarkers (K-ARPI) has recently launched a strategy for developing novel biologic markers to identify the progression speed of AD and new drug candidates for slowing the progression by modulating immune inflammatory response in a subpopulation of AD and other NDs.
We will now describe the rationale for choosing immune inflammatory modulation as a promising therapeutic target in a subpopulation of AD and related diseases. We will also describe K-ARPI's design which aims to identify new biomarkers and determine drug responsiveness.
EVIDENCE OF NEUROINFLAMMATION IN AD AND ND
The presence of a sustained neuroinflammatory response has been considered to indicate non-specific sequences of neuronal cell death in ND.20 However, emerging evidence suggests that inflammation is not only a crucial contributor in cell death mechanisms in AD, PD, ALS, and multiple sclerosis, but also one of key modifiable factors that controls interaction between the dynamic immune system and the static central nervous system (CNS).2021 In AD, immune-inflammatory activation can facilitate and exacerbate both Aβ and neurofibrillary tangle pathology. Therefore, it is a promising target for modification of disease progression.22
Microglia are resident innate immune cells in the CNS. They originate from erythro-myeloid progenitor cells in the embryonic yolk sac that migrate to the brain at around embryonic day 10.5 in mice, after which they propagate, spread, and ramify throughout the brain parenchyma.2324 Microglia are dynamic cells25 that can refine synaptic networks,26 phagocytize apoptotic cells,27 and secrete growth factors to support neuronal survival.23 Recently, comprehensive single-cell RNA analysis of CNS immune cells in diverse NDs have revealed disease-associated microglia (DAM), a subset of microglia that show a unique transcriptional and functional signature.26 DAM are associated with AD risk genes including triggering receptor expressed on myeloid cells-2 (TREM2), a family of receptors that belong to triggering receptors expressed on myeloid cells and are required for DAM activation.26272829 Microglia can induce inflammatory responses against neurodegeneration-associated molecular patterns (NAMPs), similar to systemic macrophage response against pathogen- and damage-associated stress signals (PAMPs and DAMPs).27
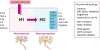
Based on the dual role of microglia in inflammation, the concept of pro-inflammatory (M1) and anti-inflammatory polarity (M2) has been established.3031 This has led to the proposal of a potential therapeutic strategy and the identification of agents that can convert M1 into an M2 phenotype.32 This would allow an optimal strategy to suppress or switch chronic inflammatory microglia to be identified. Fig. 2 schematically presents receptors known to be involved in differentiating polarity or functional phenotypes. It also shows various agents that influence characteristics of those receptors.
Fig. 2
Characteristics of microglia showing two types of polarity (M1 and M2) and their characteristic cytokines and receptors. Possible candidates potentiating M2 polarity are summarized.
M1: pro-inflammatory, M2: anti-inflammatory polarity, LPS: lipopolysaccharide, IFN: interferon, GM-CSF: granulocyte-macrophage colony-stimulating factor, TNF: tumor necrosis factor, IL: interleukin, SR: scavenger receptor, TGF: transforming growth factor, DMF: dimethylformamide, G-CSF-R: granulocyte colony-stimulating factor receptor, GSK-3: glycogen synthase kinase-3, HDAC: histone deacetylase, PPAR: peroxisome proliferator-activated receptor, AMPK: adenosine monophosphate-activated protein kinase, JAK/STAT: Janus kinase/signal transducers and activators of transcription.
The important role of neuroinflammation in cell death mechanisms of AD is supported by findings that genes encoding immune receptors of microglia including TREM2 and CD33 are associated with AD.22 The role of TREM2 in ND was originally identified in the relationship between TREM2 loss of function mutations and Nasu-Hakola disease (polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy).33 TREM2 gene is expressed in a subgroup of myeloid cells in the body, including dendritic cells, granulocytes, and tissue-specific macrophages. However, it is only expressed by microglia in the brain. In addition to its role as part of the innate immune system's response to pathogens,34 TREM2 has also been shown to be involved in phagocytosis of apoptotic neurons and clearance of aggregated proteins.31 Downregulation of TREM2 or DAP12 in microglia reduces this phagocytosis while overexpression of TREM2 has the opposite effect.35 R47H and R62H TREM2 variants have been associated with an increased risk of late-onset AD.36 Furthermore, TREM2 variant is a risk factor for FTD, PD, and sporadic ALS.37 During the preclinical period in AD, CSF levels of Aβ42 will decrease with amyloid plaque deposition in the brain. Conversely, CSF levels of tau and p-tau will increase, likely reflecting increased neuronal damage. These increases coincide with the onset of MCI and Alzheimer's dementia.38 CSF levels of soluble TREM2 (sTREM2) have been reported to be correlated with CSF levels of tau and p-tau as well as with the onset of cognitive decline.39 CSF sTREM2 levels may reflect microgliosis and inflammation related to neurodegeneration in AD pathology.40 TREM2 is protective in early stages of the disease. It facilitates clearance of intracellular and extracellular pathological tau species and damaged neuronal debris. However, it becomes pathogenic during neurodegenerative phases of the disease in which chronic inflammation, astrocytosis, and aberrant synaptic and neuronal degeneration dominate.22 In addition, during the last decade, human genetic studies, particularly genome-wide association studies using single-nucleotide polymorphisms, have identified over 20 genetic loci that are robustly associated with AD risk.4142 And, role of these genes related neuroinflammation with single or combined effects would be delineated in near future.
ALS is one of the most fatal and fast-progressing NDs. The onset and progression of inherited ALS are determined by motor neurons and microglia.43 Autopsied brain tissues in ALS have revealed microglia activation and perivascular infiltration of monocytes and T cells.44 Recently, one study has demonstrated that nuclear factor-kappa B (NF-κB) activation induces motor neuron death and reduces survival rate in a mouse model of ALS.45 In addition, clinical symptoms related to damaged upper motor neurons in patients with ALS are associated with extensive cerebral activation of microglia.46 Accordingly, replacement with healthy microglia or removal of mutant microglia can prolong survival of ALS mice.47 These findings suggest that targeting neuroinflammation processes could be further studied in clinical trials as a way to slow the progression of ALS.
To summarize the existing evidence from transgenic animal models and clinico-pathophysiological findings of AD, PD, ALS, and other NDs, all these NDs appear to be multifactorial diseases with multiple mechanisms leading to neuronal injury, although non-neuronal cells are also required for disease progression and neuronal death. This non cell-autonomous toxicity is closely related to toxic microglial activation and neuroinflammation with abnormal proteinostasis.48
NEUROINFLAMMATION RELATED DE NOVO TARGETS SUGGESTED BY THE K-ARPI
With abundant evidences suggesting the importance of immune-inflammation in pathogenic mechanisms of AD and ND, we have hypothesized that it would be valuable to develop a microglia model from peripheral blood monocyte. In our recent study, microglia like cells were induced from peripheral blood mononuclear cells by culturing them with granulocyte-macrophage colony-stimulating factor (GM-CSF) and interleukin 34 for three weeks. Their characteristic signatures were similar to brain resident microglia based on immunohistochemistry and transcriptome data as shown in Fig. 3. Transcriptome data of induced microglia like cells (induced microglia [iM]) were quite different from peripheral monocyte. This cell technology can be used as a promising tool to predict microglial status of patients in living state. However, its significance should be further validated with more detailed confirmative process.
Fig. 3
Generation of induced microglia like cells from peripheral blood mononuclear cells (A) and defected microglial phagocytosis in rapidly progressive AD compared to early AD and normal controls (B).
AD: Alzheimer's disease, GM-CSF: granulocyte-macrophage colony-stimulating factor, IL-34: interleukin-34, I-MG: induced microglia, HC: healthy controls, AD (E): Alzheimer's disease (early), AD (R): Alzheimer's disease (rapidly).

Development of novel biological markers reflecting current status of neuro-inflammatory process in the brain of AD patients or animal model would be important. Recently, sphingosine kinase 1 (SphK1) has been identified as a key factor regulating inflammatory response. SphK1 was increased in lipopolysaccharide-activated microglia. It regulates the expression of pro-inflammatory cytokines in microglia.4950 However, our data suggest that neuronal SphK1 is a main regulator of inflammatory response in AD.51 SphK1 was significantly reduced in neurons, but not in microglia of APP/PS1 mice.505152 We found that SphK1 transgenic mice exhibited widespread and high expression levels of the transgene transcript in a variety of organs and cells, including neurons and microglia of the brain.51 In contrast with elevated microglial SphK1-induced inflammation, APP/PS1/SphK1 transgenic mice showed a decrease in inflammation, although SphK1 expression was increased in microglia.51 These results suggest that neurons are the major population responsible for Aβ-mediated inflammation by SphK1, rather than microglia. In the same study, dysfunction of microglia, including an increase of pro-inflammatory markers and loss of phagocytic function, was found in AD model. Developing therapeutic agents that can restore the defective phagocytic capacity of microglia in AD may have important implications for future treatment of AD. Indeed, this is a key concept of the K-ARPI platform. Another novel interesting finding of our study was that function of SphK1 known as a “kinase” of sphingosine converting to sphinogosine-1-phosphate (S1P) was confirmed as “acetylator” of COX2. It can resolve inflammation in AD model.51 Therefore, therapeutic strategy that can increase SphK1 in AD may allow dysfunctional microglia to switch to healthy or normally functioning microglia with a normal phagocytic ability that can result in a reduction of pro-inflammatory signals and elevation of anti-inflammatory properties. The human body and cellular molecular signal pathways change dynamically according to their environment or triggering factors, including inflammation and the immune system (Fig. 4). Clinical phenotypes can manifest differently according to genetic and epigenetic changes. Polarities or functional characteristics of immune cells, including microglia and T-cells, can continuously switch according to environmental factors and body conditions (Fig. 4). Therefore, it would be important to develop therapeutic strategies that focus on switching the environment from M1 or toxic conditions to an anti-inflammatory, M2 state.
Fig. 4
Conceptual fluid-like spectra of clinical phenotypes and functional characteristics of immune cells depending on the environment.
M1: pro-inflammatory, M2: anti-inflammatory polarity, AD: Alzheimer's disease, SPM: specialized pro-resolving mediator, Th: T helper.

Another interesting finding of the on-going K-ARPI platform is that rapidly progressed AD patients' induced microglia like cells derived from patient's own peripheral monocytes show defective microglial phagocytic function with dominant M1 polarity (Fig. 4). This phenomenon was closely associated with decreased levels of m-RNA related to actin motility such as Nck-associated protein 1 (Nap1). Using these two important targets (SphK1 and Nap1), the K-ARPI has developed a research decision platform based on immune-inflammatory biomarkers. A schematic summary is shown in Fig. 5.
Fig. 5
Decision platform suggested by the K-ARPI. (A) Prescreening of AD patients for clustering with genetic information and clinical characteristics, imaging, biomarkers, and analysis. (B) Reclassification of prescreened clusters according to the algorithm using standardized biomarkers. (C) Schematic summary of the decision platform of the K-ARPI.
K-ARPI: Korean AD Research Platform Initiative based on Immune-inflammatory biomarkers, AD: Alzheimer's disease, PET: positron emission tomography, CSF: cerebrospinal fluid.

As an initial step in adopting the concept of stratification and personalized medicine, subgroups of AD patients are now clustered according to their genetic profiles, clinical characteristics, and imaging data. After creating patient clusters, their serum and CSF cytokines/chemokines related to neuroinflammation are analyzed. At the same time, de novo biomarkers of microglia, including SphK1 activity, Nap 1 expression levels, phagocytic function, and TREM2 level will be used as standardized tools for re-grouping initial clusters and determining parameters to identify candidate clusters expected to be responders to immune inflammatory modulating drugs or non-responders. In this process, tools such as deep learning programs and nomograms will be used.
CONCLUSION
Lessens from previous failed clinical trials and recent evidences associated with the importance of neuroinflammation have shown that pathophysiological mechanisms in AD are not only confined to amyloid and tau hypotheses. Neuroinflammation triggered by the CNS innate immune response may also play a central role in the pathogenesis of AD and other NDs. Moreover, immune activation could be an early cause rather than a late consequence of AD. Therefore, the development of an anti-inflammatory therapeutic strategy that potentiates protective microglia functions could be a promising model for AD treatment. We believe that the therapeutic strategy platform based on immune-inflammation modulation in AD and other NDs represents a future approach for clinical trial design and drug development.
 XML Download
XML Download