

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Prostate cancer (PCa) is the most common nonskin malignancy diagnosed in males globally [1]. More than one million patients are diagnosed with PCa yearly, and the mortality rate is estimated at over 300,000 annually [2]. Although most patients with PCa present with curable, localized disease, approximately 5% of patients will develop metastatic disease to be treated by systemic therapy, such as androgen-deprivation therapy [3]. Despite the rapid initial response to primary androgen-deprivation therapy, nearly all males with metastatic PCa eventually progress to metastatic castration-resistant PCa (mCRPC) and invariably die within 2 to 3 years [4]. There is thus an urgent need to identify novel therapeutic targets based on a better understanding of the molecular biology of metastatic PCa [5]. In this context, characterization of the genomic landscape of PCa, particularly in lethal disease, can be helpful to discover clinically actionable molecular targets.
Recent studies revealed that recurrent somatic mutations, gene fusions, copy number variation, and chromosomal alteration are significantly observed as genomic features of localized and metastatic PCa [678910]. These genomic characterizations will be useful to provide more accurate prognostic information and to predict therapeutic responsiveness using novel biomarkers. For instance, among 16 patients with mCRPC treated with a poly (ADP-ribose) polymerase inhibitor in a phase 2 trial, 14 patients with defects in DNA repair genes (BRCA2 loss and ATM aberrations) showed a high response rate to this drug [11]. However, established genomic data for metastatic PCa have mostly been derived from Western populations, in whom clinicopathologic features differ from those of other races, such as Asian populations. Actually, Korean males have more aggressive phenotypes of PCa than do Western males despite the lower incidence rate in Korean males [12].
In this context, we analyzed the mutation profiles and copy number alterations (CNAs) of Korean patients with metastatic PCa (hormone-sensitive and castration-resistant) by use of an Oncomine comprehensive assay and CNA analysis. Despite the small number of enrolled patients, this study provides crucial and novel genomic information of Korean patients with metastatic PCa based on next-generation sequencing (NGS) technology.
MATERIALS AND METHODS
1. Study population
We retrospectively reviewed the clinicopathologic data of 14 patients with metastatic PCa who were treated at our institution from 2009 through 2017 and whose tissues were evaluable for genomic analysis. The Institutional Review Board at Samsung Medical Center approved this study (approval number: 2017-03-097). Among the 14 patients with metastatic PCa, cancer tissues from 12 were retrospectively collected at the time of palliative transurethral resection of the prostate as fresh-frozen tissue and a paraffin-embedded block in our biobank facility. Conversely, we collected cancer tissues and blood samples from two patients with metastatic PCa at the time of initial prostate biopsy. We obtained written informed consents from these two patients. Using these fresh-frozen tissues, we analyzed mutation profiles and CNAs of 14 patients with metastatic PCa. To validate the results of tissue analysis, we further performed genomic analysis of two plasma samples matched with tumor tissues from prostate biopsies.
2. Oncomine comprehensive assay
DNA and total RNA were extracted by using Recover All™ Multi-Sample RNA/DNA Workflow (Invitrogen, Waltham, MA, USA) according to the manufacturer's instructions. The library was prepared by using an Oncomine Comprehensive Panel (ThermoFisher Scientific, Waltham, MA, USA) comprising 143 genes covering hotspot mutations of 73 genes, all coding exons of 26 tumor suppressor genes, copy number gains of 49 genes, and 183 fusion transcripts from 22 fusion driver genes. In that panel, DNA-specific amplicons are created using 2,530 amplicons from two primer pools, and RNA-specific amplicons are produced by using 188 unique primer pairs from one pool. Clonal amplification of the libraries was carried out by emulsion polymerase chain reaction using the Ion Chef System (ThermoFisher Scientific). The prepared libraries were then sequenced on an Ion S5 XL Sequencer using an Ion 540 Chip. Eight samples were multiplexed into one run on a 540 Chip. Data analysis was performed by using Torrent Suite v (ThermoFisher Scientific) along with the Ion Reporter v5.4 (ThermoFisher Scientific). Oncomine Comprehensive v1-540-w2.3.1-DNA and Fusions-Single Sample was used to detect low-frequency somatic variants (SNVs, indels, CNAs) and fusion. Detected variants were annotated with Oncomine Variant Annotator 5.4, which annotates a variant as a “hotspot” when it is a predefined mutation in one of 73 hotspot genes or 26 tumor suppressor genes, as “deleterious” when a variant is a novel truncating mutation in one of 26 tumor suppressor genes, as an “amplification” when the minimum ploidy gain of the 5% confidence interval is ≥2 in one of the 49 copy number gain genes, and as a “fusion” when a positive fusion call is made in one of the 194 fusion variants.
3. Low-coverage, whole-genome sequencing of PCa tissue
For low-coverage, whole-genome sequencing (LC-WGS), extracted genomic DNA was sheared to the target size (250 bp) by using Q800R (Qsonica, Newtown, CT, USA). Then, we assessed the size of the sheared genomic DNA with Tapestation 4200 (Agilent Technologies, Santa Clara, CA, USA) and constructed libraries for sequencing runs using Accel-NGS® 2S plus DNA library kits (Swift, Miami, FL, USA). We sequenced an average of 5.8 million reads from the NextSeq 500 (Illumina, San Diego, CA, USA) system using 75-bp, single-read mode. All reads were aligned to the reference genome (hg19) using the BWA-mem algorithm, and reads with a mapping quality value of 60 were selected. The reference genome was divided into 200-kilobase bins, and the number of reads was counted for each bin. The counting value of each region used the LOESS algorithm for GC correction. Using 100 healthy male genomic samples, we calculated the log2 ratio in each region using QDNA-seq R packages with default parameters. Segmentation was performed using the CBS algorithm (DNAcopy v1.38.1).
4. LC-WGS analysis of plasma samples matched with tumor tissues
Two plasma samples matched with tumor tissues (OCA13_1,13_2) were collected. We performed plasma LC-WGS for comparison of plasma and tissue chromosomal aberration patterns using the following methods. Approximately 10 mL of whole blood was collected from patients. Within 2 hours of collection, the blood samples were centrifuged at 1000×g for 15 minutes at refrigerated temperature. Plasma was transferred to microcentrifuge tubes, centrifuged at 10,000×g for 15 minutes at refrigerated temperature to exclude residual cells, transferred to fresh tubes, and stored at −80℃. For each sample, plasma circulating tumor DNA (ctDNA) was extracted from 1 mL of plasma using the QIAamp Circulating Nucleic Acid Kit from Qiagen (Hilden, Germany). The resulting ctDNA was used to make a library using a Truseq nano kit (Illumina) for sequencing, and the pooled libraries were analyzed with the NextSeq 500 (Illumina). The 75-bp, single-end reads generated from the NextSeq 500 system were mapped to hg19 reference sequences using the BWA-0.7.5a version. Duplicate reads were identified by Picard (http://broadinstitute.github.io/picard/) with the default parameters. Bias correction concerning GC contents and mappability of sequenced reads for all chromosomes was carried out using the readDepth 0.9.8.4 version of R package with default parameters. Z-scores of sequenced reads on each chromosome were compared with nine healthy male plasma samples.
RESULTS
1. Clinicopathologic and genomic characteristics of 14 patients with advanced PCa
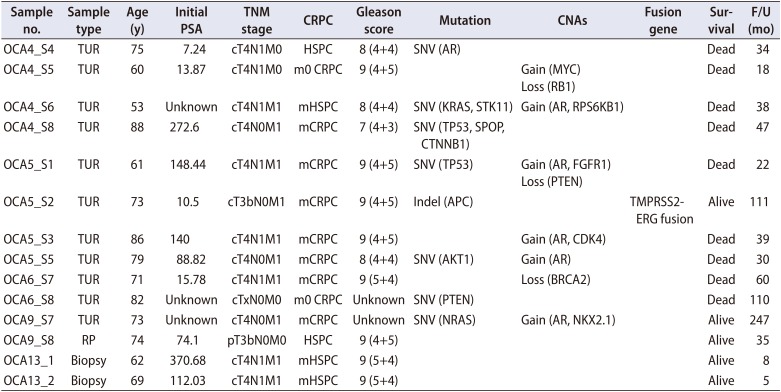
Table 1 shows the baseline clinicopathologic characteristics and genomic information from the Oncomine Comprehensive Panel of 14 patients with advanced PCa. Among 14 patients, 70% had metastatic disease and 15% had nonmetastatic CRPC and locally advanced tumors with tumor classification greater than pT3b. In patients with metastatic PCa, hormone-sensitive tumors constituted 30%, and CRPCs represented 70%. In 12 patients for whom information on Gleason scores was available, only one had a GS 7 (4+3) tumor, whereas 11 had aggressive tumors with GS 8 (25%) or GS 9 (66%).
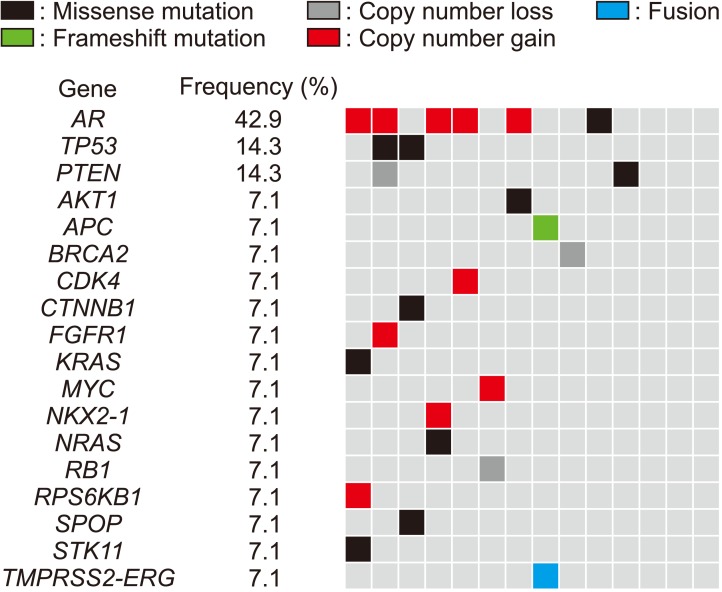
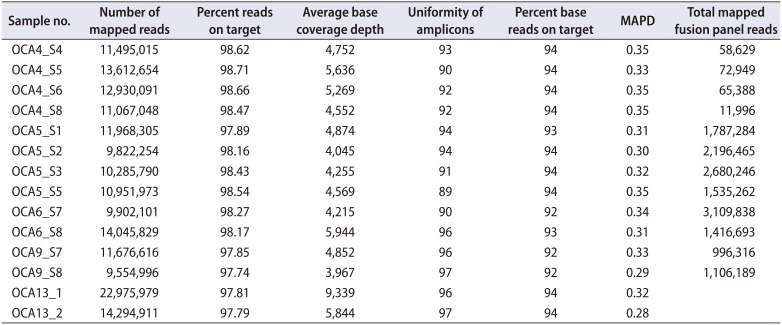
From the 14 samples of PCa tissues, all but one with a failed reading of the fusion panel passed all the quality criteria (number of mapped reads, percentage of reads on target, average base coverage depth, uniformity, percentage of base reads on target, median absolute pairwise difference [MAPD], total mapped fusion panel reads) of the Oncomine comprehensive assay (Table 2). Two samples could not be analyzed on the fusion panel. An average of 9 presumed somatic variants was detected (range, 5–16) (Supplementary Table 1). Among them, an average of 3 nonsynonymous variants were present (range, 0–5). A total of 11 known or novel mutations were detected from 8 samples (57.1%) (Supplementary Table 2). The most prevalent mutation was in the TP53 gene. One mutation in the genes SPOP, PTEN, APC, androgen receptor (AR), or CTNNB1, which are known to be associated with prostatic cancer, was detected in individual patients. Mutations in other driver genes, which were not detected in more than 1% of the samples from the TCGA dataset, were also detected (AKT1, KRAS, NRAS, and STK11 genes). A total of 13 CNAs were observed from 7 samples (50.0%) (Supplementary Table 3). The most prevalent CNA was AR amplification, which was detected in 5 samples (35.7%). Other than AR amplification, copy number loss in PTEN, BRCA2, and RB1 and copy number gain in MYC, FGFR1, MCL1, CDK4, NKX2-1, and RPS6KB1 were also detected. TMPRSS2-ERG fusion was detected in one sample (Supplementary Table 4). Excluding 3 samples (21.4%) with no aberrations on the Oncomine panel, samples had a mean of 2.3 aberrations (25 aberrations/11 samples) (Fig. 1).
2. LC-WGS analysis in tumor tissues from 14 patients with advanced PCa
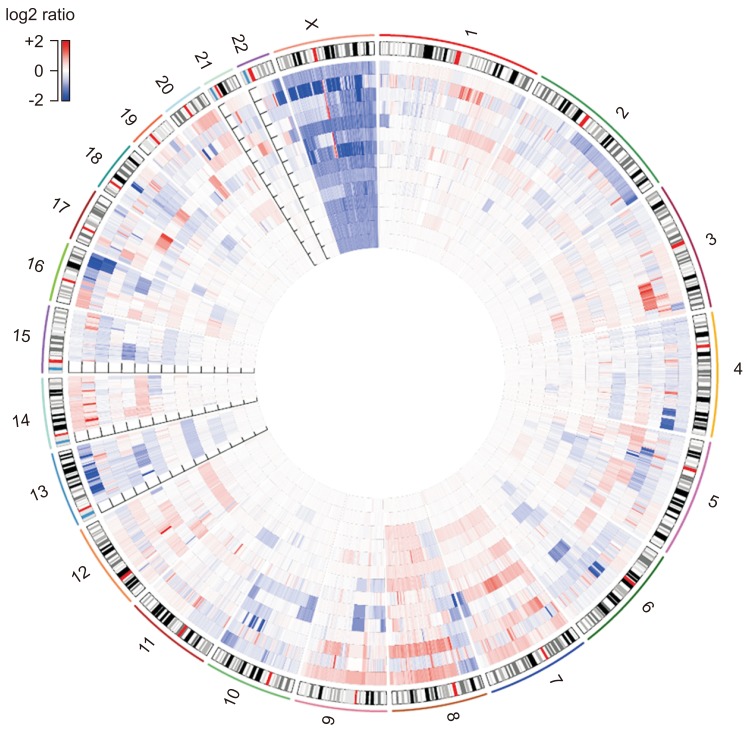
Among the 14 samples, 13 (92.9%) had detectable CNAs. CNA patterns of each sample are shown in Fig. 2. The mean patterns of copy number aberration (Supplementary Fig. 1) were similar to previously reported CNAs in PCa. Among the CNAs, the following were statistically significant when using GISTC algorithms; 8q24.21, 11q13.3, and Xq12 gains and loss of 6q16.1, 8p23.1, 10q25.1, 16q24.2 and 18q12.3, Xp22.32, Xq25 and Xq26.3 regions (Supplementary Fig. 2). All the CNAs called by the Oncomine panel were concordant in LC-WGS. One sample (OCA4_S4) showing no CNA aberration had an AR mutation.
3. CNA analysis of circulating tumor DNA in plasma samples from metastatic PCa
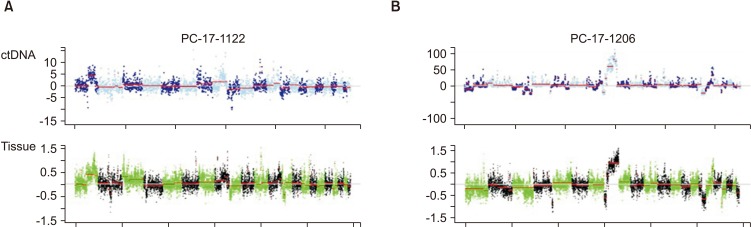
LC-WGS of two samples (OCA13_1, OCA13_2) was performed. The CNA patterns were similar between tissue and plasma (Fig. 3).
DISCUSSION
In the present study, we are the first to report the results of a genomic analysis of advanced PCa in the Korean population by using an Oncomine Comprehensive Panel and LC-WGS. We retrospectively analyzed the mutation and CNA patterns of cancer tissues. Furthermore, we analyzed two plasma samples to compare the CNA profiles between tumor DNA and circulating tumor DNA in blood. When we combined the Oncomine analysis with LC-WGS, we found at least one or more genomic alterations in all the samples. Moreover, we confirmed that CNA patterns were similar between cancer tissue and plasma from patients with advanced PCa.
For optimal treatment of advanced PCa, it is critical to understand the nature of its genetic features [13]. There have been various studies of the genomic profiles of advanced PCa in recent years [6]. A Michigan group first reported the mutational landscape of 50 patients with lethal, heavily pretreated metastatic CRPC using whole-exome sequencing [691014]. They found that nine genes were recurrently mutated, particularly AR cofactors and associated proteins, including FOXA1 and MLL2 [6]. More recently, Robinson et al. [9] reported a hallmark study of integrative genomic analysis for patients with mCRPC, performing whole-exome and transcriptome sequencing of 150 tumors. They found that TP53 and AR alterations were enriched in patients with mCRPC compared to those with only primary tumors [9]. In addition, aberrations of DNA repair genes, such as BRCA2, BRCA1, and ATM, were substantially higher than in primary tumors [9]. More interestingly, 90% of patients with mCRPC had clinically actionable aberrations, such as AR pathway alteration or PI3K and WNT signaling pathway alteration, suggesting a novel druggable target for advanced PCa in addition to AR-targeting agents [9].
In our study, the Oncomine Comprehensive Panel identified genomic aberrations in 78.6% (11/14) of the patients with PCa. The key finding of this study was that the most commonly altered genes were AR (42.9%), TP53 (14.3%), and PTEN (14.3%), which have been identified as commonly mutated in PCa. Interestingly, aberrations in AR and PTEN might have associated prognoses and therapeutics in PCa. Patients with PTEN deletion show more aggressive Gleason scores and a greater risk for recurrence of PCa [15]. AR mutations are associated with treatment responsiveness of AR-targeting agents in mCRPC [16]. In terms of types of genetic aberrations, 57.1% of patients (8/14) had SNV or indel mutations, whereas 92.9% (13/14) had CNAs observed in LC-WGS. This is consistent with previous findings that CNAs are the major alternations in PCa, as they are found in 90% of PCa tissues including primary and advanced tumors. Additionally, LC-WGS can improve the diagnostic yield of CNAs to overcome the lower detection power of the Oncomine panels.
An additional key finding of the present study was that CNA patterns in circulating tumor DNA were similar to those of tumor tissue in the LC-WGS of two samples. Of note, significant alterations of copy number were observed in patients with metastatic PCa compared to those in patients with benign prostatic hyperplasia. Thus, we believe that plasma LC-WGS can be used to monitor tissue CNAs in advanced PCa. Similar to our results, in findings from Wyatt et al. [17], all somatic mutations observed in tissue biopsies from metastatic sites were concurrently identified in circulating tumor DNA in matched plasma samples from 45 patients with mCRPC. Particularly, copy number patterns were highly correlated between PCa tissues and matched cell-free DNA in plasma samples [17]. A research group in Canada recently highlighted that circulating tumor DNA analyses could provide valuable information on the responsiveness of AR-directed therapies [18]. That group conducted whole-exome and targeted sequencing in 72 genes of cell-free DNA in blood from 202 patients with mCRPC prior to abiraterone or enzalutamide treatment [18]. Importantly, the authors found that aberrations of the DNA repair genes BRCA2 and ATM and mutations of TP53 were significantly associated with rapid resistance [18]. Because repeated tissue biopsies might be difficult due to invasiveness, liquid biopsy such as that for circulating tumor DNA can offer a promising tool for acquiring serial samples during systemic treatment and the disease course in patients with metastatic PCa [1920].
We acknowledge that there were several limitations in this study. First, a small number of patients from a single institution was the most crucial limitation in our study. Second, there were several unavoidable drawbacks of retrospective analyses, such as selection bias, heterogeneity of patients, and inconsistent sample quality. Third, in contrast with the previous results in advanced PCa, our study population had relatively small numbers of mutations per patient according to the Oncomine Comprehensive Panel. Although the actual number of mutations in our population could be lower than those in other studies, the limited coverage of genes included in the Oncomine panel could be the reason for the lower numbers of detected mutations per patient in the current study. Nevertheless, our study for the first time provides valuable insight into the genomic characteristics of Korean patients with advanced PCa.
CONCLUSIONS
In summary, the patterns of mutation and CNA in Korean patients with advanced PCa were similar to those of previous studies. In particular, at least one genomic alteration was detected in all samples by the combination of the comprehensive Oncomine panel and LC-WGS, improving the detection rate of genomic alteration. More importantly, we found that patterns in CNA in circulating tumor DNA from plasma samples were similar to those in tumor DNA from cancer tissues in patients with advanced PCa. Our study offers valuable information on the genomic features of advanced PCa in the Korean population, as well as on the potential of circulating tumor DNA as a tool for liquid biopsy in the future.

 XML Download
XML Download