

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Abstract
Over the past decade, next-generation sequencing (NGS) has evolved at an astonishing pace and has revolutionized clinical medicine as well as genomics research. The rapid advancements in NGS technologies have been accompanied by accumulating evidence of the analytical and clinical validity, and clinical utility of NGS. NGS is used worldwide. This review provides medical technicians and laboratory physicians with the essential elements for establishing clinical NGS testing. Here the authors briefly describe the advantages and drawbacks of currently available NGS platforms, potential sources of error in NGS workflow, and reference materials.
References
1. Levy SE and Myers RM. Advancements in next-generation sequencing. Annu Rev Genomics Hum Genet. 2016; 17:95–115.
2. Goodwin S, McPherson JD, McCombie WR. Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet. 2016; 17:333–51.

3. Minear MA, Alessi S, Allyse M, Michie M, Chandrasekharan S. Noninvasive prenatal genetic testing: current and emerging ethical, legal, and social issues. Annu Rev Genomics Hum Genet. 2015; 16:369–98.
4. Park KJ, Park S, Lee E, Park JH, Park JH, Park HD, et al. A population-based genomic study of inherited metabolic diseases detected through newborn screening. Ann Lab Med. 2016; 36:561–72.
5. Bodian DL, Klein E, Iyer RK, Wong WS, Kothiyal P, Stauffer D, et al. Utility of whole-genome sequencing for detection of newborn screening disorders in a population cohort of 1,696 neonates. Genet Med. 2016; 18:221–30.
6. Pritchard CC, Salipante SJ, Koehler K, Smith C, Scroggins S, Wood B, et al. Validation and implementation of targeted capture and sequencing for the detection of actionable mutation, copy number variation, and gene rearrangement in clinical cancer specimens. J Mol Diagn. 2014; 16:56–67.
7. Lih CJ, Sims DJ, Harrington RD, Polley EC, Zhao Y, Mehaffey MG, et al. Analytical validation and application of a targeted next-generation sequencing mutation-detection assay for use in treatment assignment in the NCI-MPACT trial. J Mol Diagn. 2016; 18:51–67.
8. Saunders CJ, Miller NA, Soden SE, Dinwiddie DL, Noll A, Alnadi NA, et al. Rapid whole-genome sequencing for genetic disease diagnosis in neonatal intensive care units. Sci Transl Med. 2012; 4:154ra135.
9. Segal JP. Next-generation profciency testing. J Mol Diagn. 2016; 18:469–70.
10. Aziz N, Zhao Q, Bry L, Driscoll DK, Funke B, Gibson JS, et al. College of American Pathologists'laboratory standards for next-generation sequencing clinical tests. Arch Pathol Lab Med. 2015; 139:481–93.
11. Aird D, Ross MG, Chen WS, Danielsson M, Fennell T, Russ C, et al. Analyzing and minimizing PCR amplifcation bias in Illumina sequencing libraries. Genome Biol. 2011; 12:R18.
12. Benjamini Y and Speed TP. Summarizing and correcting the GC content bias in high-throughput sequencing. Nucleic Acids Res. 2012; 40:e72.
13. Jennings LJ, Arcila ME, Corless C, Kamel-Reid S, Lubin IM, Pfeifer J, et al. Guidelines for validation of next-generation sequencing-based oncology panels: a joint consensus recommendation of the Association for Molecular Pathology and College of American Pathologists. J Mol Diagn. 2017; 19:341–65.
14. Robasky K, Lewis NE, Church GM. The role of replicates for error mitigation in next-generation sequencing. Nat Rev Genet. 2014; 15:56–62.
15. Jones S, Anagnostou V, Lytle K, Parpart-Li S, Nesselbush M, Riley DR, et al. Personalized genomic analyses for cancer mutation discovery and interpretation. Sci Transl Med. 2015; 7:283ra53.
16. Kebschull JM and Zador AM. Sources of PCR-induced distortions in high-throughput sequencing data sets. Nucleic Acids Res. 2015; 43:e143.
17. Illumina. https://support.illumina.com/content/dam/illumina-marketing/documents/products/other/miseq-overclustering-primer-770-2014-038.pdf. (updated on August 2017).
18. Pabinger S, Dander A, Fischer M, Snajder R, Sperk M, Efremova M, et al. A survey of tools for variant analysis of next-generation genome sequencing data. Brief Bioinform. 2014; 15:256–78.
19. Oliver GR, Hart SN, Klee EW. Bioinformatics for clinical next generation sequencing. Clin Chem. 2015; 61:124–35.
20. Chiara M and Pavesi G. Evaluation of quality assessment protocols for high throughput genome resequencing data. Front Genet. 2017; 8:94.
21. Gargis AS, Kalman L, Bick DP, da Silva C, Dimmock DP, Funke BH, et al. Good laboratory practice for clinical next-generation sequencing informatics pipelines. Nat Biotechnol. 2015; 33:689–93.
22. Santani A, Murrell J, Funke B, Yu Z, Hegde M, Mao R, et al. Development and validation of targeted next-generation sequencing panels for detection of germline variants in inherited diseases. Arch Pathol Lab Med. 2017; 141:787–97.
23. Hardwick SA, Deveson IW, Mercer TR. Reference standards for next-generation sequencing. Nat Rev Genet. 2017; 18:473–84.
24. Schmitt MW, Kennedy SR, Salk JJ, Fox EJ, Hiatt JB, Loeb LA. Detection of ultra-rare mutations by next-generation sequencing. Proc Natl Acad Sci U S A. 2012; 109:14508–13.
25. Sims DJ, Harrington RD, Polley EC, Forbes TD, Mehaffey MG, McGregor PM 3rd, et al. Plasmid-based materials as multiplex quality controls and calibrators for clinical next-generation sequencing assays. J Mol.
26. Kudalkar EM, Almontashiri NA, Huang C, Anekella B, Bowser M, Hynes E, et al. Multiplexed reference materials as controls for diagnostic next-generation sequencing: a pilot investigating applications for hypertrophic cardiomyopathy. J Mol Diagn. 2016; 18:882–9.
27. Illumina. https://www.illumina.com/content/dam/illumina-marketing/documents/products/technotes/hiseq-phix-control-v3-technical-note.pdf. (updated on August 2017).
28. Duncavage EJ, Abel HJ, Merker JD, Bodner JB, Zhao Q, Voelkerding KV, et al. A model study of in silico profciency testing for clinical next-generation sequencing. Arch Pathol Lab Med. 2016; 140:1085–91.
29. Davies KD, Farooqi MS, Gruidl M, Hill CE, Woolworth-Hirschhorn J, Jones H, et al. Multi-institutional FASTQ fle exchange as a means of profciency testing for next-generation sequencing bioinformatics and variant interpretation. J Mol Diagn. 2016; 18:572–9.
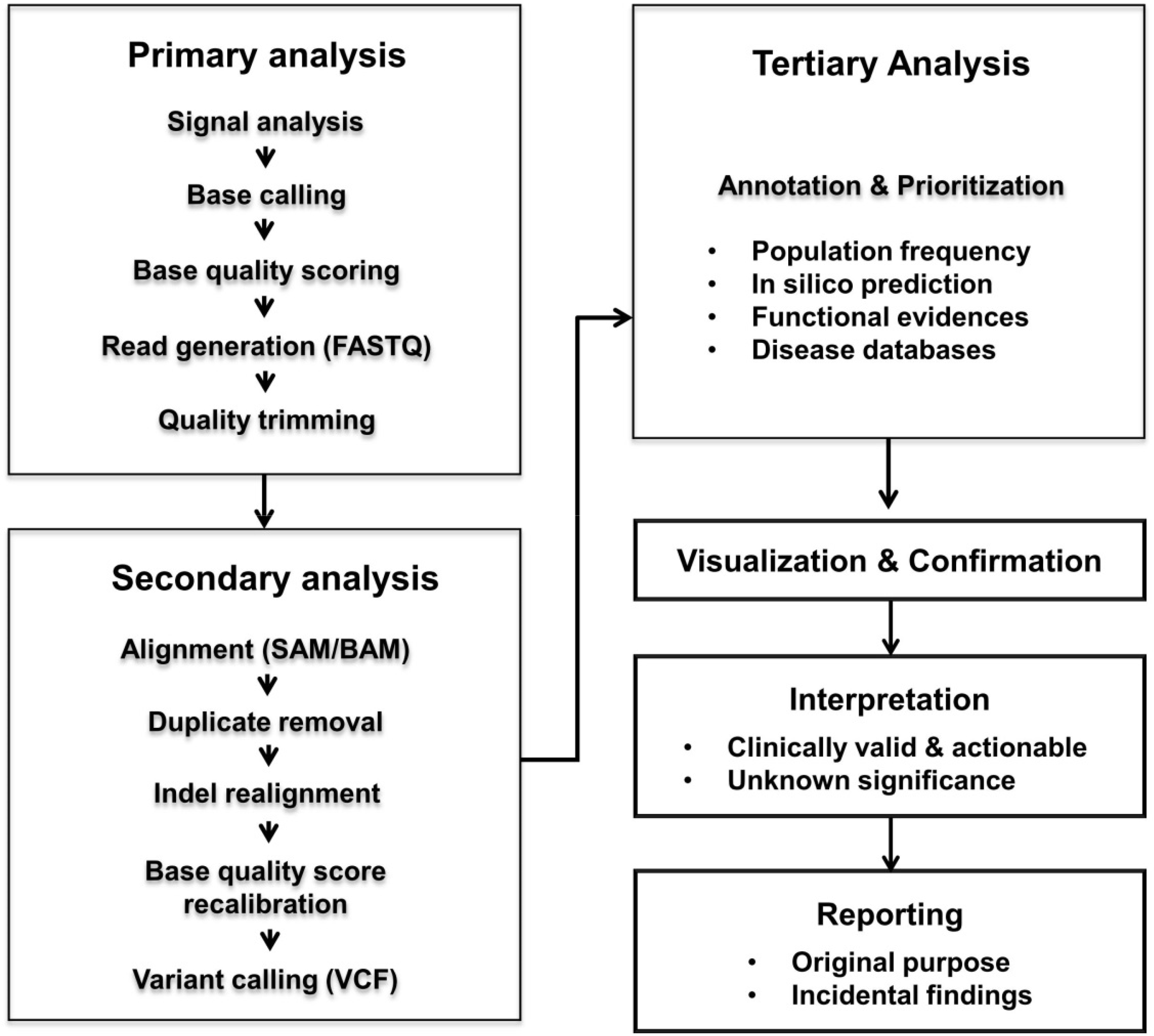
Table 1.
Summary of major next-generation sequencing
Table 2.
Potential sources of error and quality control
| Error sources∗ | Quality control and optimization† | |
|---|---|---|
| Sample preparation | ∙ User errors (mislabeling) | ∙ Specimen rejection criteria according to sample types |
| ∙ Low yield of nucleic acid | ∙ OD 260/280, OD 230/280 | |
| ∙ Nucleic acid contamination (microorganism, xenograft) | ∙ DNA/RNA integrity number | |
| ∙ Low tumor cell fraction (heterogeneity) | ∙ DNA integrity (gel image) | |
| ∙ Nucleic acid degradation (FFPE) | ∙ Tumor cell fraction (depending on depth of coverage) | |
| Library construction | ∙ User errors (carry-over, contamination) | ∙ Size and concentration of fragmented DNA |
| ∙ Adapter dimers | ∙ Size distribution of library | |
| ∙ Index swab | ∙ Library quantification and normalization | |
| ∙ Low library complexity, PCR duplication | ∙ Size selection | |
| ∙ Capture bias (GC contents, repeating elements, pseudogenes, etc.) | ∙ Length and match of index and barcode | |
| ∙ Primer dimer, amplification errors, allele dropout | ∙ Primer/probe: tiling design or rebalancing | |
| ∙ Flow cell overloading/underloading | ∙ Cluster density | |
| Sequencing | ∙ Decline in signal intensity | ∙ Base call quality scores (Q-score) |
| ∙ Incorrect base incorporation | ∙ Per base GC content | |
| ∙ Sequence context (GC contents, homologous gene, homopolymers) | ∙ Sequence length distribution | |
| ∙ Platform-specific error | ∙ Duplicate sequence | |
| ∙ Per base N contents | ||
| ∙ Ti/Tv ratio | ||
| Bioinformatic analyses | ∙ Low depth of coverage | ∙ On-target coverage |
| ∙ Uneven of coverage | ∙ Mapping rate | |
| ∙ Misalignment | ∙ Mapping quality | |
| ∙ Strand bias | ∙ Confirmation with orthogonal methods | |
| ∙ Low concordances among different bioinformatics tools | ∙ False positive and false negative rates | |
| ∙ Quality trimming | ||
| ∙ Duplicate removal | ||
| ∙ Local realignment | ||
| ∙ Base quality score recalibration | ||
| ∙ Variant quality score recalibration |
Table 3.
Comparison of target enrichment methods
 XML Download
XML Download