

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Helicobacter pylori is a gull-wing-shaped, Gram-negative, microaerophilic bacterium with pathogenic virulence factors including urease, flagella, and colistin [1]. In recent years, meaningful progress has been made in detecting H. pylori in the mouth, and several studies using different detection methods have shown that the oral cavity is a secondary location for H. pylori colonization [2]. Krajden et al. [3] first discovered the existence of H. pylori strains in oral plaque biofilm in 1989. Previous studies have indicated that H. pylori, especially in subgingival plaque, is associated with periodontitis. Dye et al. [4] reported that even after adjusting for socioeconomic factors, the presence of H. pylori and periodontal disease were significantly correlated. In individuals with periodontitis, H. pylori was detected more frequently in plaque than in healthy people [567]. Moreover, the H. pylori detection rate in subgingival plaque may be positively correlated with the severity of periodontitis, suggesting that H. pylori could be a risk factor for chronic periodontitis [46]. Periodontal disease is a complex, multifactorial disease that occurs in periodontal tissue [8], and most studies on the causes of periodontal diseases have mainly focused on the dominant bacteria responsible for periodontal diseases, such as Porphyromonas gingivalis [9]. As a result, some potential periodontal pathogenic bacteria that are difficult to cultivate but are involved in periodontal inflammation have been neglected.
There is evidence that the periodontal pocket is a nutrient-rich environment that is conducive to H. pylori colonization [5]. In subgingival plaque, H. pylori is protected from external environmental factors, such as the host's immune defense mechanisms, as well as from potentially toxic substances, such as acids and antibiotics. The subgingival environment also facilitates nutrient uptake, co-existence with other species, and removal of potentially harmful metabolic substances [10]. The diversity of microbial species in the periodontal ecological environment and the persistent inflammatory environment may provide a favorable pathway for the establishment of bacterial flora [11]. For example, experiments have shown that Campylobacter spp. and Fusobacterium spp. can coaggregate with H. pylori. It is possible that the colonization and adhesion of H. pylori in the oral cavity may be facilitated by interactions with these oral microflora [121314].
Although epidemiological studies of H. pylori in the oral cavity have been performed, none have explored the effects of H. pylori infection on periodontitis and investigated the infection mechanism. Human periodontal ligament fibroblasts (hPDLFs) are important in the repair of periodontal inflammation, and the quality of the collagen they produce can affect the repair process and the structure and function of periodontal tissue. In a preliminary experiment, we established an optimal model of the infection of hPDLFs by H. pylori SS1, in which invasion was observed by transmission electron microscopy. In this study, the effect of H. pylori on the proliferation and cell cycle of hPDLFs was evaluated using the Cell Counting Kit-8 (CCK-8) method, Ki-67 immunofluorescence, and flow cytometry. Additionally, the transcription and protein expression levels of cell cycle-related factors were measured, with the aim of clarifying the effect of H. pylori SS1 on hPDLFs.
MATERIALS AND METHODS
Bacterial strains
A standard strain of H. pylori (H. pylori SS1) was donated by Professor Yong Sun of the Digestive Department of Nanfang Hospital. The strain was subjected to 16S rDNA sequencing identification, and cultured on Columbia agar plates containing 6% fresh defibrinated sheep blood in a microaerobic atmosphere (7.2% CO2, 6% O2, 7% H2, and 79.7% N2) with 90% humidity in an anaerobic jar (Anoxomat mark II, MART Microbiology, Lichtenvoorde, the Netherlands) at 37°C. After culture in the microaerobic environment for 2 days, bacteria were collected from the plate for experiments or stored at −80°C.
Cell culture
All samples were healthy premolars that needed to be removed for treatment purposes, taken from patients (14 to 25 years old) at Nanfang Hospital. The protocol of this study was approved by the medical ethics committee of Nanfang Hospital, Southern Medical University, Guangzhou, China (NFEC-2017-017). Patients were informed of the sample acquisition and provided written informed consent. Periodontal ligaments were collected to culture periodontal fibroblasts. Cells were cultured to passages 3–8 for experiments.
Establishment of an H. pylori invasion model in vitro
According to the results of our preliminary experiment conducted to determine the optimal co-culture conditions for an invasion model, H. pylori SS1 was co-cultured at different multiplicities of infection (MOIs) (MOI=0, 10:1, 100:1) with hPDLFs for 6 hours, and the cells were then washed 6 times with phosphate-buffered saline (PBS) (Gibco, Invitrogen, Mulgrave, Australia) containing gentamicin (100 µg/mL) and incubated with gentamicin-containing (100 µg/mL) culture medium for 2 hours to kill extracellular H. pylori SS1.
Electron microscopic observation of H. pylori invasion of cells
After the invasion model (MOI=100:1) was constructed, the cell morphology was observed under an inverted microscope and photographed. The cells were washed with PBS 3 times, scraped off, collected, and centrifuged before being fixed with 2.5% glutaric dialdehyde. The samples were rinsed, fixed, dehydrated, embedded in sections, and stained. Finally, the samples were observed and photographed under a transmission electron microscope.
Cell proliferation detected by the CCK-8 method and immunofluorescence of the nuclear antigen Ki-67
The hPDLFs were digested, resuspended, and cultured in 96-well plates (1×103 cells/well) and 24-well plates (1×104 cells/well). The cells were divided into 3 groups (MOI=0, 10:1, 100:1). The corresponding amount of bacteria (determined by a spectrophotometer) was added, followed by culturing for 6 hours and incubation with gentamicin-containing (100 µg/mL) culture medium. CCK8 kits (Dojindo, Kumamoto, Japan) were used to detect cell proliferation for 7 consecutive days on the 96-well plates. After the 6-hour invasion model of H. pylori SS1 infection was established, cells in the 24-well plates were fixed with 4% formalin for 20 minutes and permeated with 0.2% Triton X-100 (Leagene, Beijing, China), followed by blocking with serum and incubation with Ki-67 and DAPI antibodies (ABclonal, Woburn, MA, USA).
Cell cycle analysis
After establishing the H. pylori SS1 invasion model with different concentrations, cells were digested and resuspended, washed twice with PBS, and converted to a single-cell suspension (1×105 cells/mL). Next, 70% cold ethanol was added to fix the cells, followed by storage at −4°C overnight. Cells were washed with PBS before dyeing, the supernatant was removed, and PI/RNase A dyeing work fluid (Keygentec, Nanjing, China) was added, followed by incubation in the dark for 30 minutes. Cells were analyzed with flow cytometry, the data were processed using specialized online software (Diva 7.0, Becton Dickinson, Franklin Lakes, NJ, USA), and the proportion of cells in each cell cycle stage was calculated.
Quantitative real-time polymerase chain reaction
Total RNA was isolated with a total RNA extraction kit (Sangon, Shanghai, China) and reverse-transcribed to cDNA with a PrimeScript RT Reagen Kit (Takara, Shiga, Japan). Following the manufacturer's instructions, SYBR Premix Ex Taq (Takara) was used for the quantitative analysis of real-time polymerase chain reaction with the following primers: cyclin B1: F5′-ATGCAGCACCTGGCTAAGAA-3′, R5′-TACACCTTTGCCACAGCCTT-3′; cell division cycle 25C (Cdc25C): F5′-ACTGCCACTCAGCTTACCAC-3′, R5′-AGCTGTGCTGGGCTACATTT-3′; glyceraldehyde-3-phosphate dehydrogenase (GAPDH): F5′-GTCAAGGCTGAGAACGGGAA-3′, R5′-AAATGAGCCCCAGCCTTCTC-3′; cyclin dependent kinase 1 (CDK1):F5′-CTGGCTGATTTTGGCCTTGC-3′,R5′-GAGTAACGAGCTGACCCCAG-3′. The reaction conditions were as follows: 40 cycles at 95°C for 10 seconds, 60°C for 20 seconds, and 72°C for 20 seconds. The data were analyzed using the 2−ΔΔCt method.
Western blotting
The cytoplasmic protein lysate was extracted from sodium dodecyl sulfate buffer and then separated into equal amounts for sodium dodecyl sulfate-polyacrylamide gel electrophoresis. The proteins were transferred to a polyvinylidene difluoride membrane (GE Healthcare, Marlborough, MA, USA), and the membranes were blocked for 1 hour (dissolved in TBST). Then, the proteins were probed with primary antibodies for Cdc25C, serine phosphorylation of Cdc25C (Cdc25C-S216), CDK1, tyrosine phosphorylation of CDK1 (CDK1-Y15), cyclin B1, and GAPDH overnight at 4°C. The membranes were incubated with the secondary antibodies for 1 hour. Finally, an electrochemiluminescence detection system (Modular E170, Roche Diagnostics, Basel, Switzerland) was used and the net optical density of the strip was analyzed using Image-Pro Plus 6.0.
Statistical analysis
The results were analyzed using SPSS 19.0 (IBM Corp., Armonk, NY, USA). Data are shown as the mean±standard deviation. The 2-tailed Student t-test or 1-way analysis of variance was used to examine differences between groups, and P values <0.05 were considered to indicate statistically significant differences.
RESULTS
H. pylori SS1 invaded hPDLFs and was localized in the cytoplasm
After H. pylori SS1 co-culture with cells for 6 hours, we observed that some coccoid forms of H. pylori SS1 were localized in the cytoplasm. The ultrastructure showed clear increases in the number of intracellular vacuoles. Furthermore, H. pylori SS1 appeared to be surrounded by vacuoles in the cytoplasm, where it could proliferate and divide (Figure 1B and C).
Figure 1
The ultrastructure of hPDLFs was observed by transmission electron microscopy. (A) Control at ×5,000 magnification. (B) Invasive group at ×6,000 magnification. The black arrow indicates that H. pylori is surrounded by cytoplasmic vacuoles. (C) Invasive group at ×20,000 magnification. The black arrow indicates that H. pylori is surrounded by cytoplasmic vacuoles. The yellow arrow indicates the proliferation and division of H. pylori in cytoplasmic vacuoles.
hPDLFs: human periodontal ligament fibroblasts.

H. pylori SS1 inhibited the proliferation of hPDLFs
Compared with the control samples, cell proliferation decreased to a certain extent in the samples co-cultured with H. pylori SS1. After 6 hours of co-culture, the proliferation capacity of cells decreased according to both the CCK-8 method (Figure 2A) and the Ki-67 immunofluorescence results (Figure 2B). In the first 3 days, cell proliferation was suppressed, but without statistical significance. However, from day 4, the proliferation of hPDLFs gradually started to show significant decreases (P<0.05, P<0.01 in the low- and high-concentration groups, respectively) and significantly lower proliferation was observed in the high-concentration group (MOI=100:1).
Figure 2
Effects of H. pylori infection on the proliferation of hPDLFs. (A) OD values of hPDLFs infected by H. pylori for 7 consecutive days. (B) Immunofluorescence staining for the Ki-67 protein in hPDLFs.
hPDLFs: human periodontal ligament fibroblasts, OD: optical density; MOI: multiplicity of infection.
a)Statistically significant difference (P<0.05); b)Statistically significant difference (P<0.01).

G2 phase arrest after H. pylori infected
After the H. pylori SS1 invasion models of hPDLFs were established (MOI=0, 10:1, 100:1; co-culture for 6 hours), the proportion of DNA in each cell cycle stage was measured by flow cytometry (Figure 3A-C). As the H. pylori SS1 concentration increased, the proportion of cells in the experimental group in the G1 phase gradually decreased, while the proportion of cells in the G2 phase gradually increased (Figure 3D and E) compared to those in the control group.
Figure 3
The proportion of DNA content in cell cycle stages in hPDLFs was measured by flow cytometry. (A-C) The proportion of DNA content in various stages of the cell cycle. (D) Change in the proportion of cells in various stages of the cell cycle. (E) Change in the proportion of cells in the G2 phase.
hPDLFs: human periodontal ligament fibroblasts; MOI: multiplicity of infection.
a)Statistically significant difference (P<0.01).

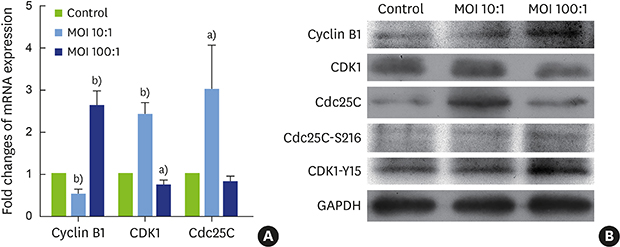
Changes in mRNA transcription levels of cell cycle-related factors in the G2 phase
As H. pylori SS1 was found to induce G2 phase arrest, we then assessed its effect on G2 phase cell cycle regulators, including the cyclin B1–CDK1 complex and Cdc25C (Figure 4A). In the low-concentration group (MOI=10:1), transcription of cyclin B1 was inhibited, while mRNA levels of CDK1 and Cdc25C were significantly higher than in the control group. However, in the high-concentration group (MOI=100:1), the expression of CDK1 was significantly lower (P<0.05), while transcription of cyclin B1 was promoted (P<0.01).
Figure 4
Changes in the levels of mRNA transcription and protein expression of cell cycle-related factors in G2 phase. (A) mRNA expression levels of cyclin B1, CDK1, and Cdc25C. (B) Protein expression levels of cyclin B1, CDK1, CDK1-Y15, Cdc25C, and Cdc25C-S216.
CDK1: cyclin dependent kinase 1, Cdc25C: cell division cycle 25C, CDK1-Y15: tyrosine phosphorylation of CDK1, Cdc25C-S216: serine phosphorylation of Cdc25C, MOI: multiplicity of infection, GAPDH: glyceraldehyde-3-phosphate dehydrogenase.
a)Statistically significant difference (P<0.05); b)Statistically significant difference (P<0.01).

Changes in protein expression levels of cell cycle-related factors in the G2 phase
In hPDLFs, the protein expression of Cdc25C and CDK1 in the low-concentration group increased, while in the high-concentration group, CDK1 protein expression decreased and Cdc25C protein increased slightly. The expression of cyclin B1 decreased in the low-concentration group and increased in the high-concentration group, roughly consistent with the mRNA results. Cdc25C-S216 and CDK1-Y15 protein expression increased after infection (Figure 4B).
DISCUSSION
Periodontal disease is a complex, multifactorial disease that occurs in periodontal tissue, which can even become the focus of infection, leading to a range of systemic diseases. Many studies have shown that H. pylori is closely related to periodontal diseases [151617]. The periodontal pocket environment is suitable for H. pylori survival, as the inflammatory environment of persistent infection in periodontal pockets provides nutrients. However, the role of H. pylori in the development of periodontal disease remains unclear. Therefore, we aimed to establish an optimal model of periodontal fibroblast invasion in vitro. In a preliminary gentamicin protection experiment, different concentrations (MOI=0, 10:1, 100:1) of H. pylori SS1 were co-cultured with hPDLFs and counted after cells were isolated from lysed cells. The optimal invasion model in our pre-experiment involved 6 hours of co-culture, and subsequent experiments were conducted using this experimental model.
According to a large number of studies on H. pylori and related gastrointestinal diseases, H. pylori is a special intracellular pathogen that can invade cells and complete the entire biological cycle through cell division and replication [18]. Intracellular invasion by H. pylori may be dependent on receptor-mediated endocytosis and tyrosine kinase [19]. After H. pylori invasion, intracellular phagocytic vacuoles are formed, which can leave cells under appropriate conditions, causing the cycle of infection to repeat. We found that H. pylori SS1 invaded hPDLFs, implying that this process may be a potential threat for periodontal disease induction and chronic infection. According to previous studies, H. pylori has a complex mechanism of cell invasion and survival in vivo [20212223]. When in an inhospitable environment, H. pylori seeks refuge in host cells [24] and transforms into its coccoid form, while preserving basic metabolic functions and some crucial structures such as the flagella [25]. In the early stages of infection, H. pylori SS1 must adapt to the internal environment of the host and complete self-proliferation or morphological changes according to the environment, leading to a slight decrease in hPDLF proliferation. Moreover, the host cell may initially activate the corresponding defense mechanisms to remove the bacteria, with only a slight effect on cell proliferation. However, the results of subsequent experiments proved that H. pylori SS1 has inhibitory effects on hPDLF proliferation. The proliferation ability of hPDLFs could be affected by the secretion of virulence proteins or toxic substances such as urease, oxidase, and catalase by H. pylori SS1. Furthermore, H. pylori SS1 may adapt to the internal environment of the host cell and escape from host defense substances, after which it may accelerate its multiplication, thereby affecting the proliferation function of host cells.
When facing infection, the G2 checkpoint may lead to changes in regulatory proteins relevant to the cell cycle in response to DNA damage, preventing the cell from entering mitosis [26]. We found that hPDLFs were arrested in the G2 phase after H. pylori SS1 infection, and demonstrated changes in the transcription and expression of cycle-related factors. The cyclin B1–CDK1 complex is known to be the key regulator of the G2 phase, and requires that cyclin B1 bind to CDK1. In the low-concentration group, the decrease in cyclin B1 may have caused failure of the formation of its complex with CDK1, resulting in G2 phase arrest. In contrast, in the high-concentration group, the transcription and protein expression levels of cyclin B1 were upregulated, while CDK1 was suppressed. This was probably due to blocking of cyclin B1 nuclear transport. There are 42 residues at the amino terminus of cyclin B1 that are implicated in its nuclear transport. Phosphorylation of the serine residues on the cytoplasmic retention sequence causes cyclin B1 to remain in the cytoplasm instead of undergoing nuclear transport [27].
The nuclear redistribution of these proteins is an important switch in the regulation of mitosis [28]. Furthermore, the phosphorylation of proteins affects their activity and changes their rate of nuclear import and export. The activity of the cyclin B1–CDK1 complex is regulated by a complex, multistep feedback loop that involves inhibition of the phosphorylation of amino acid residues of CDK1 (Thr14 and Tyr15) mediated by the balance between the WEE family kinases and cell division cycle 25 phosphatases [2629]. After activation, the cyclin B1–CDK1 complex in turn promotes Cdc25C activation, forming a positive feedback loop [4]. During mitosis, activation of Cdc25C requires phosphorylation or dephosphorylation at multiple sites (such as dephosphorylation at Ser216) and isolation from 14-3-3 proteins [30]. In our results, levels of both 5c5C-S216 and CDK1-Y15 increased after H. pylori SS1 infection. The accumulation of Cdc25C-S216 prevents the cyclin B1–CDK1 complex from activating due to failure of dephosphorylation of CDK1 at Tyr15. The accumulation of phosphorylated CDK1-Y15 leads to continued inactivation of the cyclin B1–CDK1 complex, and ultimately G2 arrest. Phosphorylation of Cdc25C at S216 also keeps Cdc25C in the cytoplasm, preventing it from translocating to the nucleus.
Various pathophysiological phenomena can result in the deregulation of proliferation by affecting cell cycle progression. Our results demonstrate for the first time that hPDLFs respond to DNA damage caused by H. pylori SS1 infection through the Cdc25C/CDK1/cyclin B1 signaling cascade, resulting in G2 phase arrest and inhibition of cell proliferation. Interestingly, different concentrations of H. pylori SS1 had different effects on cell cycle-related mRNA and protein expression. However, the G2 phase blockade effect was also dependent on the protein phosphorylation level and nuclear transport, which will require further in-depth exploration. Likewise, the damage caused by H. pylori to periodontal tissue and the potential mechanisms of periodontitis occurrence and development still require further exploration.
 XML Download
XML Download