

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
The incidence of cardiovascular disease (CVD), the leading cause of death globally, continues to rise as life expectancy increases.1)2) Cellular senescence is linked to the onset and the progression of CVD. Various stimuli, including DNA damage and oxidative stress, have been reported to trigger cellular senescence.3) Moreover, clinical risk factors such as inflammation, hypertension, and obesity were reported to accelerate vascular aging, so-called early vascular aging.4) Senescent cells show shortened telomere length and reduced proliferation although they maintain cellular viability.5) Senescent cells are phenotypically characterized by enlarged and flattened cell morphology and accumulated senescence associated β-galactosidase (SA-β-gal).6) Furthermore, SA-β-gal-positive cells express different sets of genes, such as p53 and p16,7) which are negative regulators of the cell cycle and also serve as markers of cellular senescence.
Maladaptive activation of the renin-angiotensin system (RAS) has been shown to play a critical role in the development of CVD of different etiologies including hypertension8) and diabetes.9) Angiotensin II (Ang II) is a potent systemic vasoconstrictor and is involved in several vascular pathologies by promoting pathologic hypertrophy, fibrosis, extracellular matrix deposition and inflammation via Ang II type 1 receptor (AT1R).10)11)12) There is some evidence suggesting the role of Ang II in vascular senescence13)14) and the protective effect of RAS inhibition against it.14)15)16) AT1R blockers (ARBs) are mainly used for the treatment of hypertension and have been reported to exert pleiotropic effects to protect against oxidative and inflammatory actions.17)18) Fimasartan, developed by Boryung Pharmaceutical Co., Ltd. (Seoul, Korea), is an ARB with a selective AT1R blocking effect.19) Recent studies have reported the pleiotropic effect of fimasartan beyond blood pressure lowering in myocardial infarction20) and inflammation.21)
CYR61 is an important downstream molecule of AT1R.22) CYR61 is a cysteine-rich angiogenic protein 61 that is the first member of the CCN family (CCN1) of secreted extracellular matrix proteins in mammals.23) CYR61 was originally cloned as an immediate early gene expressed in fibroblasts after growth factor stimulation.24) CYR61 expression has been reported to be associated with vascular restenosis, angiogenesis, and tumor growth.25) Also, we previously showed that CYR61 blocking inhibits vascular smooth muscle cell (VSMC) proliferation and neointimal hyperplasia.26)
Therefore, in this study, we evaluated cellular senescence following Ang II. We hypothesized that Ang II induced VSMC senescence by regulating the expression of CYR61. We also evaluated the protective role of ARB, fimasartan, against vascular senescence.
METHODS
Reagents and antibodies
Ang II was purchased from Sigma-Aldrich (Merck Millipore Corporation, Darmstadt, Germany). PD98059, ERK1/2 inhibitor, and SB203580, p38 MAP kinase inhibitor, were purchased from Invitrogen (Carlsbad, CA, USA). Fimasartan, ARB, was provided by Boryung Pharmaceutical Co., Ltd (Seoul, Korea). Primary antibodies against p53, p16 and β-actin antibodies were purchased from Santa Cruz Biotechnology (Dallas, TX, USA), and Phospho-ERK1/2, ERK1/2, Phospho-p38 MAPK and p38 MAPK antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). CYR61 antibody was purchased from Abcam. Secondary antibodies were purchased from Santa Cruz Biotechnology (Dallas, TX, USA).
Cell culture and adenoviral vectors
Human coronary artery smooth muscle cells (hCSMCs) were purchased from Lonza (CC-2583, Basel, Switzerland). Six- to eight-passage hCSMCs were cultured in SMC growth medium consisting of basal media, insulin, human recombinant epidermal growth factor, human recombinant fibroblast growth factor, gentamicin, amphotericin, and 5% fetal bovine serum at 37°C in an atmosphere of 95% air and 5% CO2 based on manufacturer's recommendations. Recombinant adenoviral vectors, expressing CYR61 cDNA (Ad-CYR61) and antisense CYR61 cDNA (Ad-AS-CYR61) fragments, were used for the overexpression or suppression of CYR61 gene in cultured hCSMCs. As a control, an adenoviral vector expressing green fluorescence proteins only (Ad-GFP) was used.
SA-β-gal staining assay
Cellular senescence was examined by SA-β-gal activity using Senescence Detection Kit (Calbiochem, Merck Millipore Corporation, Darmstadt, Germany). Briefly, cultured hCSMCs were washed with 1× PBS and fixed with Fixative Solution at room temperature for 15 minutes, and washed with PBS again. Then fixed cells were incubated with Staining Solution Mix at 37°C overnight. Cellular images were acquired with a fluorescence microscope (DM 5500B, LEICA Microsystems, Milton Keynes, UK) using a digital imaging system (DFC360FX, LEICA). SA-β-gal positive cells were counted per 50 cells and normalized to vehicle treated cells.
Total RNA isolation and quantitative real-time polymerase chain reaction (PCR)
Changes in CYR61 gene expression were determined by quantitative real-time PCR. Cultured hCSMCs were washed twice with PBS and cell pellets were collected for RNA isolation. And total RNA was isolated from the cell pellets by the RNeasy mini Kit (Qiagen, Hilden, Germany) and reversely transcribed using the amfiRivert cDNA Synthesis Premix (GenDEPOT, Katy, TX, USA) based on manufacturer's instructions. Quantitative PCR performed using SYBR Green PCR kit (Applied Biosystem, Foster City, CA, USA) and StepOnePlus Real-Time PCR System (Applied Biosystem). Real-time PCR was performed with glyceraldehyde 3-phosphate dehydrogenase (GAPDH) primers (forward: 5′-GGA AGG TGA AGG TCG GAG TC-3′, reverse: 5′-GAA GGG GTC ATT GAT GGC AAC-3′), CYR61 primers (forward: 5′-TCT CGT TGC TGC TCA TGA AAT T-3′, reverse: 5′-TAG AGT GGG TAC ATC AAA GCT TCA G-3′), p53 primers (forward: 5′-GCC CAA CAA CAC CAG CTC CT-3′, reverse: 5′-CCT GGG CAT CCT TGA GTT CC-3′) and p16 primers (forward: 5′-CCC AAC GCA CCG AAT AGT TA-3′, reverse: 5′-ACC AGC GTG TCC AGG AAG -3′). All genes were normalized to the GAPDH mRNA level.
Western blot assay
Cultured hCSMCs with the indicated reagents were washed twice with PBS and collected. Cell pellets were lysed in RIPA cell lysis buffer (Santa Cruz Biotechnology), and protein concentration was determined by BCA protein assay (Thermo Scientific Pierce, Rockford, IL, USA). The 30 µg of proteins were loaded and separated on 8% or 10% sodium dodecyl sulfate-polyacrylamide gels and transferred to activated polyvinylidene fluoride membranes (Immobilon-P; Merck Millipore, Darmstadt, Germany) by methanol. The transferred membranes were blocked with blocking buffer (5% skim milk in TBST) at room temperature for 60 minutes and incubated with the indicated primary antibodies in blocking buffer overnight at 4°C. The membranes were washed three times with TBST and incubated at room temperature with secondary antibodies conjugated to horseradish peroxidase at 1:2,500 for 60 minutes in TBST with 2% skim milk and washed three times again. Proteins were detected using enhanced chemiluminescence detection reagents (Promega, Madison, WI, USA) and the protein levels were acquired through densitometric scanning. Obtained values were expressed in arbitrary densitometric units and normalized to those of β-actin to correct for total protein loading.
Statistical analysis
All data were presented as mean±standard deviation. Statistical analyses were performed with GraphPad Prism (GraphPad Software, Inc., La Jolla, CA, USA) using the student's t-test for comparing 2 groups or one-way analysis of variance followed by the Turkey post hoc test for comparing >2 groups. A probability value of less than 0.05 was considered statistically significant.
RESULTS
Ang II induces Ang II-induced cellular senescence in hCSMCs, whereas fimasartan inhibits it
We treated hCSMCs with 1 to 100 nM of Ang II and evaluated cellular senescence by counting SA-β-gal-positive cells. Ang II treatment for 7 days significantly increased SA-β-gal-positive cells at 10 nM (2.63±0.75-fold) and at 100 nM (3.31±0.26-fold) compared with the control (Figure 1A). In contrast, Ang II-induced SA-β-gal activity (5.77±1.05-fold vs. the control) was significantly inhibited by pretreatment with 1 μM of fimasartan (2.0±0.53-fold vs. the control with Ang II, Figure 1B).
Figure 1
Ang II induces cellular senescence in hCSMCs, whereas Fima inhibits it. (A) Response of SA-β-gal positive cells following treatment with Ang II (1–100 nM) for 7 days. (B) Cellular senescence induced by Ang II was near completely blocked by pretreatment with ARB, Fima (1 µM). Scatter plots with bar show data from 4 independent experiments. Values are given as mean±standard deviation (n=4).
Ang II = angiotensin II; ARB = Ang II type 1 receptor blocker; Fima = fimasartan; hCSMC = human coronary artery smooth muscle cell; SA-β-gal = senescence-associated β-galactosidase.
*p<0.01, †p<0.0001.

Next, we evaluated the expression levels of cell senescence regulators, p53 and p16 following Ang II treatment by western blot analysis and Real-time PCR.13) Both p53 and p16 protein expressions were significantly increased (p53: 1.39±0.17, p16: 1.19±0.10-fold vs. the control, Figure 2A and Supplementary Figure 1) and their mRNA levels were similarly increased (p53: 3.20±0.78, p16: 7.79±1.01-fold vs. the control, Figure 2B), while these Ang II-induced increases were completely inhibited in both protein (p53: 1.02±0.12, p16: 0.96±0.07-fold vs. the control, Figure 2A and Supplementary Figure 1) and mRNA (p53: 0.15±0.13, p16: 0.27±0.22-fold vs. the control, Figure 2B) by fimasartan.
Figure 2
Ang II induces p53 and p16 expression, whereas Fima inhibits it. hCSMCs were treated with Ang II at 100 nM for 4 hours, and/or pretreated Fima at 1 μM for 2 hours. (A) Western blot and (B) real-time PCR analyses showed expression of p53 and p16 after Ang II treatment. Scatter plots with bar show data from 3 independent experiments. Values are given as mean±standard deviation (n=3).
Ang II = angiotensin II; Fima = fimasartan; hCSMC = human coronary artery smooth muscle cell; PCR = polymerase chain reaction.
*p<0.001, †p<0.0001.

These observations suggest that Ang II promotes the hCSMCs senescence via accumulated stress through the AT1R-p53/p16 dependent pathway, and fimasartan blocks the AT1R to regulate hCSMCs senescence.
Ang II-induced CYR61 promotes cellular senescence via AT1R-p53-dependent pathway
It was previously reported that the expression of CYR61 is induced by Ang II, and CYR61 was reported to induce cellular senescence in fibroblasts.27) Therefore, we investigated whether CYR61 mediates Ang II-induced cellular senescence in hCSMCs. First, we performed quantitative real-time PCR to assess the CYR61 gene transcription on Ang II-treated hCSMCs. The CYR61 mRNA expression levels were increased in Ang II-treated hCSMCs over 10 nM (10 nM: 1.52±0.27, 100 nM: 1.54±0.28-fold vs. the control, Figure 3A). Ang II rapidly induced CYR61 mRNA expression within 30 minutes (1.5±0.34-fold vs. the control) and peaked at 120 minutes (3.1±0.80-fold vs. the control, Figure 3B). Conversely, Ang II-induced CYR61 mRNA expression (1.55±0.36-fold. vs. the control) was completely inhibited by fimasartan (0.95±0.23-fold vs. the control, Figure 3C).
Figure 3
Ang II induces CYR61 mRNA expression in hCSMCs, whereas Fima inhibit it. CYR61 mRNA expression was detected by quantitative real-time PCR. (A) CYR61 mRNA expression was increased by following Ang II treatment in concentration-dependent manners (1–100 nM, 120 minutes). (B) Increased CYR61 mRNA expression induced by Ang II at 100 nM was continued for 240 minutes and peaked at 120 minutes. (C) Increased expression of CYR61 mRNA induced by Ang II at 100 nM for 120 minutes was significantly inhibited by pretreated Fima (1 μM). Bar graphs show data from 3 independent experiments. Values are given as mean±standard deviation (n=3).
Ang II = angiotensin II; AT1R = angiotensin II type 1 receptor; CYR61 = cysteine-rich angiogenic protein 61; Fima = fimasartan; GAPDH = glyceraldehyde 3-phosphate dehydrogenase; hCSMC = human coronary artery smooth muscle cell.
*p<0.05, †p<0.01.

We investigated whether CYR61 directly induces hCSMCs senescence. For the overexpression or the suppression of CYR61, we used a replication adenovirus encoding CYR61-specific cDNA (Ad-CYR61) or CYR61-specific antisense cDNA (Ad-As-CYR61) fragments. In CYR61-overexpressed hCSMCs, SA-β-gal (+) cells were significantly increased (3.47±0.65-fold vs. the control transfected with Ad-GFP, Figure 4A). Conversely, when CYR61 expression was blocked with Ad-AS-CYR61, Ang II-induced SA-β-gal (+) cell numbers were significantly decreased (Ang II-treated hCSMC with Ad-GFP: 3.73±0.23 and Ang II-treated hCSMC with Ad-As-CYR61: 1.77±0.60-fold vs. the vehicle-treated hCSMC with Ad-GFP, Figure 4B).
Figure 4
CYR61 regulates cellular senescence in hCSMCs. hCSMCs were transfected with adenoviral vectors at 50 MOI for 4 hours for induction or suppression CYR61, and control were transfected with Ad-GFP. (A) Induction of CYR61 by transfection with Ad-CYR61 significantly increased SA-β-Gal (+) hCSMCs. (B) However, suppression of CYR61 by transfection with Ad-As-CYR61 decreased SA-β-Gal (+) cells after Ang II treatment (100 nM). Scatter plots with bar show data from 4 independent experiments. Values are given as mean±standard deviation (n=4).
Ad-CYR61 = adenoviral vectors expressing CYR61; Ad-As-CYR61 = adenoviral vectors expressing antisense CYR61; Ad-GFP = adenoviral vector expressing green fluorescence proteins; Ang II = angiotensin II; CYR61 = cysteine-rich angiogenic protein 61; hCSMC = human coronary artery smooth muscle cell; SA-β-Gal = senescence-associated β-galactosidase.
*p<0.001, †p<0.0001.

p16 and p53 were reported to be involved in different pathways of cellular senescence.7) Therefore, we examined whether CYR61-induced hCSMC senescence was related to the p16- or p53-dependent pathway. In CYR61-overexpressed hCSMCs, expression of CYR61 and p53 was increased, whereas p16 expression was not changed in protein (CYR61: 37.94±3.26-fold, p53: 1.57±0.16-fold, p16: 0.95±0.10-fold vs. the control transfected with Ad-GFP, Figure 5A, and Supplementary Figure 2A-C) and mRNA (CYR61: 3.23±0.60-fold, p53: 11.90±1.18-fold, p16: 1.03±0.40-fold vs. the control transfected with Ad-GFP, Figure 5B). In addition, the p53 expression by CYR61 was increased in a dose-dependent manner (25 MOI: 1.36-fold, 125 MOI: 1.42-fold, 250 MOI: 1.71-fold vs. the control transfected with Ad-GFP, Figure 5C and Supplementary Figure 2D and E). However, in CYR61-suppressed hCSMCs using Ad-As-CYR61, induction of p53 by Ad-CYR61 or Ang II was significantly suppressed, whereas p16 expression was not changed in both condition (Figure 5D and E and Supplementary Figure 2F-K).
Figure 5
Ang II-CYR61 dependent cellular senescence was mediated by the p53-dependent pathway, but not by the p16-dependent pathway. hCSMCs were transfected with the indicated adenoviral vectors for 4 hours. Control were transfected with Ad-GFP. (A, B) CYR61 and p53 expressions were increased in transfection with Ad-CYR61 (50 MOI). In contrast, p16 expression was not changed. Scatter plots with bar show data from 4 independent experiments. Values are given as mean±standard deviation (n=4). (C) CYR61-induced p53 expressions were increased in a MOI-dependent manner. (D, E) CYR61 inhibition by Ad-AS-CYR61 (50 MOI) significantly inhibited p53 expression, whereas no significant change was observed in p16 expression.
Ang II = angiotensin II; Ad-CYR61 = adenoviral vectors expressing CYR61; Ad-AS-CYR61 = adenoviral vectors expressing antisense CYR61; Ad-GFP = adenoviral vector expressing green fluorescence proteins; CYR61 = cysteine-rich angiogenic protein 61; hCSMC = human coronary artery smooth muscle cell; ns = not significant.
*p<0.01, †p<0.001.

These results showed that Ang II induced CYR61 via AT1R and CYR61 mediated Ang II-induced hCSMCs senescence. Also, CYR61-induced cellular senescence was mediated by the p53-dependent pathway, not by the p16-dependent pathway.
Ang II induces CYR61 expression through the ERK/p38 MAPK signaling pathway
Lastly, we investigated the mechanisms of CYR61-associated Ang II dependent hCSMCs senescence in more detail. It was previously reported that Ang II-related cellular senescence was contributed to by MAPK, such as ERK1/2 and p38 MAPK.28) Based on these reports, we investigated the role of ERK/p38 MAPK pathways in CYR61-mediated hCSMCs senescence. First, we evaluated ERK1/2 and p38 MAPK activation by measuring phosphorylation in Ang II-treated hCSMCs. As expected, the phosphorylation levels were both increased by Ang II, and blocked by fimasartan pretreatment (Figure 6A and Supplementary Figure 3A and B). Next, we evaluated CYR61 expression in the presence of either PD98059, an ERK1/2 inhibitor, or SB203580, a p38 MAPK inhibitor, in Ang II-stimulated hCSMCs. Ang II increases CYR61 mRNA expression level (2.06±0.30-fold vs. the control, Figure 6B), and both inhibitors completely inhibited CYR61 expression mRNA level (ERK inhibitor: 0.92±0.29-fold, p38 MAPK inhibitor: 0.59±0.19-fold vs. the control, Figure 6B). Immunoblot analysis showed that the inhibition of ERK1/2 by PD98059 attenuated the expression of both CYR61 and p53 but not p16, whereas p38 MAPK inhibition by SB203580 attenuated CYR61 and p16 but not p53 (Figure 6C and Supplementary Figure 3C-G). These data showed that CYR61 was downstream of ERK1/2 and p38 MAPK, and directly controlled p53 expression, but not p16 in Ang II-induced hCSMCs senescence.
Figure 6
p53 expression of Ang II-induced senescent hCSMCs was through ERK/p38 MAPK/CYR61 signaling pathway. (A) Western blot analysis shows that Ang II (100 nM, 240 minutes) induced the phosphorylation of ERK1/2 and p38 MAPK, which was inhibited by ARB, Fima (1 μM). (B) Ang II-induced CYR61 mRNA expression is mediated by both ERK1/2 and p38 MAPK. Scatter plot with bar shows data from 3 independent experiments. Values are given as mean±standard deviation (n=3). (C) Ang II-induced activation of ERK and p38 MAPK are inhibited by PD98059 (20 μM, 20 minutes) and SB203580 (10 μM, 20 minutes), respectively, but ERK1/2 was activated by SB203580. CYR61 expression is attenuated both by PD98059 and SB203580, whereas p53 expression levels are suppressed only by PD98059, whereas p16 expression level was decreased only by SB203580, respectively.
Ang II = angiotensin II; ARB = Ang II type 1 receptor blocker; CYR61 = cysteine-rich angiogenic protein 61; Fima = fimasartan; hCSMC = human coronary artery smooth muscle cell.
*p<0.01, †p<0.001.

The proposed signaling pathways of Ang II-induced hCSMCs senescence were summarized in Figure 7.
Figure 7
The proposed signaling pathways of Ang II-induced hCSMCs senescence. ARB, Fima, may contribute to anti-senescence effects by inhibiting ERK/p38 MAPK/CYR61/p53 signaling pathway.
Ang II = angiotensin II; ARB = Ang II type 1 receptor blocker; CYR61 = cysteine-rich angiogenic protein 61; Fima = fimasartan; hCSMC = human coronary artery smooth muscle cell.
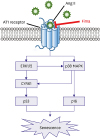
DISCUSSION
In this study, we demonstrated that Ang II induced hCSMC senescence via AT1R, which was protected against by fimasartan. We showed that CYR61 plays an important role in Ang II-induced cellular senescence in hCSMCs. Ang II, through activating ERK1/2, p38 MAPK, p16 and p53, induces vascular senescence, and CYR61 is mediates between ERK1/2 and p53. Therefore, ERK1/2-CYR61-p53 signaling axis may be a crucial pathway regulating Ang II-induced cellular senescence (Figure 7).
Vascular aging is characterized by changes in vascular structure and functions including intimal thickening and medial stiffness.29) VSMCs are the primary cell type in the tunica media, and the status of VSMCs can influence the structure and the function of blood vessels. Previous reports suggested several mechanisms for the promotion of stress-induced vascular cell senescence by Ang II. For example, Ang II activates intracellular reactive oxygen species generation-mediated nicotinamide adenine dinucleotide phosphate (NADPH) oxidase. NADPH, in turn, activates several MAPKs such as ERK1/2 and p38 MAPK, thus promoting vascular senescence.28) Also, oxidative stress induced by Nox1-based NADPH oxidase plays important roles in Ang II-induced cellular senescence.30)
Here, we showed that Ang II significantly induced hCSMC senescence via CYR61 expression and ARB, fimasartan, near completely inhibited CYR61-mediated cellular senescence. Given these findings, we proposed that Ang II induces hCSMC senescence via accumulated stress through CYR61 and ERK/p38 MAPK/p53 signaling pathway, and AT1R blocking effectively regulates VSMC senescence.
The main purpose of this study was to investigate Ang II-dependent vascular aging mechanism and anti-senescent effect of fimasartan. We clearly showed fimasartan completely blocks Ang II-induced senescence in hCSMCs. Moreover, we showed that CYR61 has important role in previously known Ang II-induced cellular senescence mechanisms. However, in order to clinically apply these results, we need to confirm the role of CYR61 and effect of fimasartan in animal experiments. Therefore, in future studies, it is necessary to confirm the role of CYR61 and the effect of fimasartan using the Ang II-induced animal aging model.
In conclusion, we showed that Ang II induced hCSMC senescence via CYR61 and ERK/p38 MAPK/p53 signaling pathway, and fimasartan suppressed Ang II-induced hCSMC senescence. These results provide the evidence that blockade of CYR61 may contribute to suppression of vascular aging and CVD.
 XML Download
XML Download