

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Congenital analbuminemia (CAA) is an autosomal recessive disease characterized by extremely low levels of serum albumin without specific causes that can induce hypoalbuminemia (i.e., reduced protein synthesis due to hepatic dysfunction or re-distribution of protein aside from loss of protein from blood vessels in the kidneys or gastrointestinal tract). Since the absence of key serum proteins in adults can be partially offset by increased synthesis of other serum proteins, this condition is usually considered to be benign. Therefore, patients with CAA typically only exhibit very mild clinical symptoms, such as fatigue, ankle edema, and hypotension.1234
CAA diagnosis relies on the elimination of other clinical conditions that can lead to hypoalbuminemia and sequencing of the ALB gene to detect mutations. In all reported cases, a mutation in ALB located on chromosome 4 (4q11-13) has been identified as a driver of CAA and is induced by homozygous or heterozygous DNA deficiency.5
CASE REPORT
In January 2018, a 28-year-old Korean male visited Department of Endocrinology, Seoul National University Bundang Hospital because of hypoalbuminemia, hypoproteinemia, hypocalcemia, and vitamin D deficiency that had been detected during a health check-up. The patient had not been experiencing physical discomfort. There was no relevant personal or family medical history. Height and weight were 170 cm and 63 kg. Physical examination revealed a blood pressure of 107/70 mm Hg, pulse rate of 70 per minute, respiration of 18 per minute, and body temperature of 36.8℃. Cardiovascular, respiratory, abdominal, and neurological assessments produced normal outcomes.
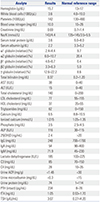
Blood test outcomes indicated normal complete blood counts and electrolytes. In addition, liver function test and thyroid function test outcomes were normal. Blood abnormalities were hypoproteinemia, hypoalbuminemia, moderate hypocalcemia, vitamin D deficiency, and elevated parathyroid hormone (PTH) and alkaline phosphatase (ALP) levels.
We performed kidney function, complement, spot urine, and albuminuria tests to eliminate the possibility of nephrotic syndrome, and the outcomes were normal. All laboratory test outcomes are summarized in Table 1. A kidney ultrasound examination revealed kidneys of normal size with normal parenchyma. Esophagogastroduodenoscopy outcomes were normal, and the patient did not exhibit suspicious symptoms of protein loss (e.g., diarrhea). The patient showed elevated PTH levels and underwent a parathyroid scan, which proved normal. Dual energy X-ray absorptiometry confirmed a normal level of bone density: Z-scores were −1.0 [bone mineral density (BMD) 0.703 g/cm2] on the left femoral and −0.8 (BMD 0.858 g/cm2) on the spine (Fig. 1).
Serum protein electrophoresis was performed to detect low albumin concentrations and compensatory increases of non-albumin proteins, especially α1 and α2 globulin fractions (Table 1). To confirm the diagnosis of CAA at the molecular level, we performed ALB mutation analysis. DNA sequencing revealed a novel mutation of heterozygous single nucleotide polymorphism (SNP) causing C>T transition at position c.1668 C>T, p.Leu556= in the ALB gene (Fig. 2).
Daily oral consumption of calcium 500 mg and vitamin D 1000 IU was commenced to treat hypocalcemia and vitamin D deficiency. At an outpatient follow-up visit after 3 months, the physical examination outcomes were still normal. Blood 25(OH)D levels had increased from 2.4 to 19.3 ng/mL. PTH levels had decreased from 234 to 141 pg/mL, and ALP levels had decreased from 116 to 104 IU/L. Throughout five outpatient follow-up visits, serum albumin concentrations remained below 2.2 g/dL, and no specific clinical symptoms or signs were evident.
This study was approved for exemption of subject consent by Seoul National University Bundang Hospital Institutional Review Board (IRB No. B-1905/538-701).
DISCUSSION
The diagnosis of CAA is typically based on hematological indices, serum protein electrophoresis, and genetic analysis. Serum albumin concentrations can vary from 1 to 10 g/dL.1 CAA is extremely rare, with approximately 70 cases reported worldwide. The cases are constantly being updated in the albuminemia registry.5 Thus far, all molecular level studies on CAA have indicated that a mutation in the ALB gene near the centromere of chromosome 4 (4q11-13, 74269972-74287129) is the driver of this disease. The ALB gene is located on chromosome 4 and is divided into 15 exons by 14 introns.6 Identification of various mutations causing the onset of CAA suggests that the disease is genetically heterogeneous.56
CAA is a risk factor of high morbidity and mortality during pregnancy and infancy, indicating that albumin plays a critical role in the prenatal and perinatal periods.78 However, since a low level of serum albumin can be partially supplemented by increased levels of other serum proteins, CAA in adults does not result in clear symptoms and, thus, is considered a relatively benign disease. The benign features of CAA often lead to a misdiagnosis or delayed diagnosis of this rare condition.9
Serum albumin is a key transporter of serum calcium. Approximately 45% of circulating calcium is attached to serum albumin.2 It is expected that a patient with low serum albumin concentrations will exhibit low levels of total serum calcium despite having normal levels of biologically active ionized calcium.2 In addition, serum albumin acts as a transporter of vitamin D. The majority of 25(OH)D and 1,25-Dihydroxyvitamin D circulating in the bloodstream is tightly bound to the vitamin D binding protein, 10–15% is bound to albumin, and <1% of circulating vitamin D exists in an unbound form.10 Therefore, hypoalbuminemia may have effects on vitamin D deficiency. Nonetheless, hypocalcemia and vitamin D deficiency are thought to rarely affect the bone density of healthy young males (as in this case report), although they may have more severe effects in different age groups.78
The only limitation of this study was that the medical history of family members could only be assessed via question-answer interviews. We could not confirm the history of hypoalbuminemia in the patient's family members, and the presence of genetic mutations could not be assessed.
The 28-year-old Korean male patient harbored a heterozygous SNP c.1668C>T, p.Leu556= in the ALB gene. The patient presented with hypoalbuminemia with accompanying hypocalcemia and vitamin D deficiency, but no other clinical symptoms. Continued interest in this rare disease and efforts in genetic diagnosis will contribute to uncovering the molecular genetic basis of CAA.


 XML Download
XML Download