

 PDF
PDF ePub
ePub Citation
Citation Print
Print



Introduction
Idiopathic pulmonary fibrosis (IPF) is the irreversible form of the idiopathic interstitial pneumonia that has symptoms of chronic and progressive dyspnea by unknown causes12. IPF is etiologically characterized with alveolar epithelial cell injury, fibroblast/myofibroblast proliferation, and excessive extracellular matrix (ECM) deposition leading to the destruction of the alveolar structure. Recently, the incidence and prevalence of IPF have been increased with emerging medical attention in public. In addition, environmental factors including stone, sand, silica, cigarette smoke and wood/metal/micro dust are seriously considered to IPF associated risk factors345. Those risk factors cause the release of pro-inflammatory cytokines, ECM deposition leading to irreversible alveolar destruction6. Representatively, pirfenidone and nintedanib have been evaluated in clinical trials of patient with IPF due to anti-fibrotic effect, however, these therapeutics exhibit the context of certain limitations regarding safety and specific efficacy78. Although the pathogenesis of IPF which is associated with progressive fibro-proliferative as well as destructive pathway of alveolar epithelial cell, the underlying mechanism is still unknown.
Bleomycin (BLM) is a glycopeptide antibiotic with potent tumor therapeutic in various squamous carcinoma of testicular and lymph nodes. However, its adverse effect has been associated with lung injury. BLM-induced pulmonary toxicity is chemotoxic related to pulmonary fibrosis with symptoms of irregular respiration and cough910. In this aspect, the animal and cellular models of BLM induced lung fibrosis injury have been widely used due to the cytotoxic effects. In pulmonary parenchyma of IPF, alveolar epithelial cells have been considered as primary damaged cells of surrounding inflammatory11. Sisson et al.12 has been reported that alveolar epithelial type II cells (AE II) in diphtheria toxin-exposed mouse lung functionally inhibited fibroblast proliferation and collagen synthesis. Moreover, BLM has promoted the release of proinflammatory cytokines such as tumor necrosis factor α (TNF-α), interleukin-6 (IL-6), and transforming growth factor β1 (TGF-β1)13. It has been generally accepted that fibrotic diseases are promoted by TGF-β1 and appear AE II cells apoptosis, accumulation of collagen and ECM, and fibroblast proliferation1014. Emodin, an antibacterial anthraquinone exhibited the down-regulation of anti-apoptotic protein Bcl2 and up-regulation of pro-apoptotic protein BAX promoted apoptosis in TGF-β1-stimulated human fibroblasts15. In addition, BLM-induced DNA double-strand breaks increased expression of the p21/p53 pathway that involved cell cycle arrest and cell apoptosis16. Recently, Schlafen (SLFN) family are proteins that have biological functions the effect that regulation of lymphocyte differentiation, growth and T-cell development/activation17. It has been reported that SLFN family have regulation of cell cycle progression and growth arrest1819. Baicalein (Bai) induced G1 cell cycle arrest and apoptosis in osteosarcoma cell20. Moreover, Bai induced G1/G2 cell cycle arrest in rat heart endothelial cells21. We used Bai in order to inhibit G1/G2 cell cycle of MLE-12 cells. Therefore, in this study, we determined whether SLFN family is involved in the regulation of cell proliferation and apoptosis in BLM or Bai exposed mouse AE II (MLE-12 cells).
Go to : 
Materials and Methods
1. Chemicals
BLM was purchased from Tokyo Chemical Industry (#B3972; Tokyo, Japan). Methylthiazolyldiphenyl-tetrazolium bromide (MTT) were bought from Sigma-Aldrich (#M5655; St. Louis, MO, USA). Hoechst 33258 were bought from Sigma-Aldrich (#94403). Bai was purchased from Sigma-Aldrich (#465119).
2. Cell culture and treatment
MLE-12 cells (#CRL-2110; American Type Culture Collection, Manassas, VA, USA) maintained in DMEM/F-12 (#SH30023.1; HyClone, South Logan, UT, USA) with 2% fetal bovine serum (FBS; #FBSUS500-S; AusGeneX, CA, Santa Clara, USA), 1% penicillin/streptomycin, 0.005 mg/mL insulin, 30 nM Sodium selenite, 10 nM Hydrocortisone, and 10 nM β-estradiol and incubated in incubator (37℃, 95% air/5% CO2). MLE-12 cells maintained FBS-free media at 12 hours before BLM treatment. After starvations, BLM treated dose of 1–10 µg/mL for 24 hours in MLE-12 cells. Bai dissolved in dimethyl sulfoxide (DMSO) and treated dose of 0.01–100 µM for 24 hours in MLE-12 cells.
3. MTT assay
MLE-12 cells were grown in a 96-well plate and it was treated BLM or Bai for 24 hours. After treatment, 20 µL/mL of MTT reagent were added 100 µL per well for 2 hours at 37℃. The reagent was removed and each well was added 100 µL of DMSO and dissolved purple formazan crystals. The measurement was wavelength 570 nm of absorbency using microplate reader (BioTek, Winooski, VT, USA). Calculation of cell viability was divided BLM treatment group into non-treatment.
4. Quantitative real-time reverse transcription-polymerase chain reaction
Total RNA from MLE-12 cells was isolated using Trizol Isolation Reagent (#79306; Qiagen, NRW, Düsseldorf, Germany). For reverse transcription-polymerase chain reaction, 1 µg of total RNA was reverse transcribed for cDNA synthesis in each tube using Revers Transcription-premix (#EBT-1514; Elpis Biotech, Daejeon, Korea). Quantitative real-time reverse transcription-polymerase chain reaction (qRT-PCR) was used with SYBR Green (#RT501; Enzynomics, Daejeon, Korea) for gene expression of mRNA levels. Two pairs of primers are shown in Table 1. Relative quantification of gene expression was normalized Ct values of β-actin each sample and calculated by 2−ΔΔCt methods.
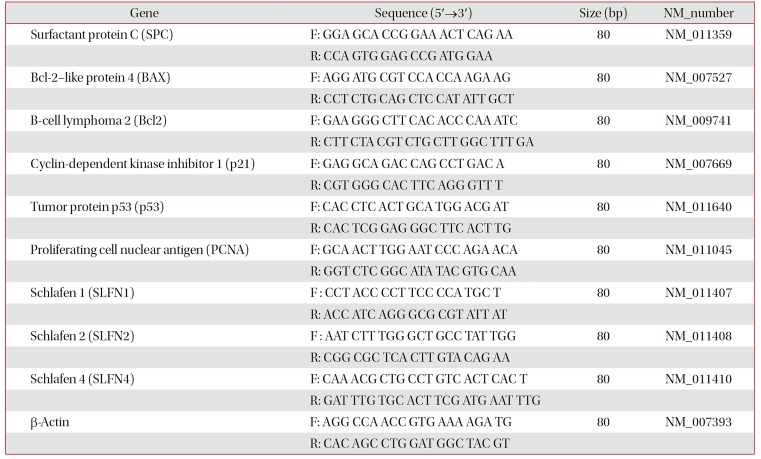
Table 1
Sequences of the primers in the qRT-PCR analysis
![]()
5. Enzyme-linked immunosorbent assay
After BLM treatment in MLE-12 cells for 24 hours were measured TNF-α (#DY410), IL-6 (#DY406), and TGF-β1 (#DY1679) cytokine levels. Measurement of cytokine level was used DuoSet enzyme-linked immunosorbent assay kit (R&D Systems, Minneapolis, MN, USA) in MLE-12 cell culture supernatant. All data were obtained duplicated experiments repeated at least three.
6. Hoechst 33258 staining
MLE-12 cells were grown at on sterile cover glass and treated BLM for 24 hours. The culture medium was removed through suction and the cells were washed that used with phosphate buffered saline. The cells were fixed with 4% paraformaldehyde for 10 minutes at room temperature (RT) and permeabilized with 0.2% Tripton X-100 for 10 minutes at RT. The cells were incubated with 1 µg/mL Hoechst 33258 for 10 minutes at 37℃ and were mounted on the slide. Photographs were observed using a fluorescence microscope (Olympus, Tokyo, Japan). The abnormal index was measured by and calculated percentages (%).
7. Immunocytochemistry
MLE-12 cells were grown at on sterile cover glass and treated BLM for 24 hours. The cells were fixed with 4% paraformaldehyde for 10 minutes, permeabilized with 0.2% Tripton X-100 for 10 minutes. The cells were stained nuclei with DAPI (#F6057; Sigma-Aldrich). Photographs were studied using a confocal laser-scanning microscope (#LSM 510; Carl Zeiss, Stuttgart, Germany). The nuclear size was obtained by nuclear area measured using ImageJ.
8. Propidium iodide staining
The cells were grown at 6-well plate and treated BLM for 24 hours. The cells harvested and fixed in 70% ethanol for 15 minutes at −20℃. DNA staining was using propidium iodide (PI) staining reagent (#550825; BD Sciences, San Jose, CA, USA) for 15 minutes at RT. Results were analyzed using Flow Cytometry (Accuri C6; BD Biosciences).
9. Statistical analysis
All results were shown as the mean±standard deviation. One-way analysis of variance (ANOVA) was calculated for comparisons of multiple groups and Dunnett's test was used by post hoc test. Statistically significant was considered the value of p<0.05, p<0.01, and p<0.001. All results were analyzed using Prism 5 and repeated experiments at least three.
Go to :
Results
1. BLM increased pro-inflammatory cytokines with loss of cell viability
In order to determine whether BLM affects cellular proliferation, MLE-12 cells were treated with BLM (1–10 µg/mL) for 24 hours. The morphological change occurred flatten and enlargement of cell body in MLE-12 cells exposed to BLM under the microscope (Figure 1A). In MTT assay, cell viability was significantly decreased in treatment up to 10 µg/mL BLM (Figure 1B). In qRT-PCR analysis, mRNA level of surfactant protein C (SPC) which is a specific marker of lung AE II cell was decreased (Figure 1C). Moreover, pro-inflammatory cytokines including TNF-α, IL-6, and TGF-β1 were significantly released in response to BLM treatment in MLE-12 cells (Figure 1D–F).
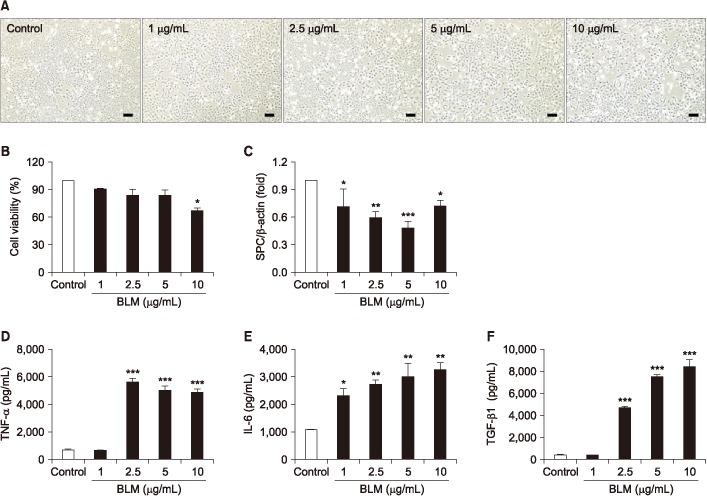 | Figure 1In the MLE-12 cells was evaluated toxicity effects of by bleomycin (BLM). MLE-12 cells were treated BLM (1–10 µg/mL) for 24 hours. (A) MLE-12 cells were induced morphological change by BLM. Scale bars=100 µm. (B) MLE-12 cells were treated with BLM and determined cell viability using MTT assay. (C) Expression of alveolar epithelial cell marker, surfactant protein C (SPC) was measured by quantitative real-time reverse transcription-polymerase chain reaction. The graph was measured inflammatory cytokines of tumor necrosis factor α (TNF-α) (D), interleukin-6 (IL-6) (E), and transforming growth factor β1 (TGF-β1) (F) in cell culture supernatant in enzyme-linked immunosorbent assay. *p<0.05, **p<0.01, ***p<0.001.
|
2. BLM-induced apoptosis and cell cycle arrest with nuclear enlargement
We determine whether BLM induces apoptosis and cell cycle arrest in MLE-12 cells. In Hoechst 33258 staining, the nucleus of BLM treated MLE-12 cells were occurred condensation and fragmentation in a dose-dependent manner under the microscope (Figure 2A). Moreover, in qRT-PCR analysis, BLM treatment increased mRNA expression of a pro-apoptotic factor (BAX) while decreased expression of anti-apoptotic factor (Bcl2) (Figure 2B). Cell cycle arrest was analyzed using DAPI and PI staining. In DAPI staining, cellular nucleus enlargement was morphologically found with loss of cell number (Figures 2A, 3A, B). It has been shown that increased size of nucleus is associated with cell cycle arrest leading to senescence22. Therefore, we measured nuclear size and the abnormal index. As a result, nucleus size and abnormal index were significantly increased in BLM exposed in MLE-12 cells (Table 2). In addition, in flow cytometry analysis, BLM decreased G0/G1 whereas increased S and G2/M phase in MLE-12 cells (Figure 3B). These findings suggest that BLM increased nuclear enlargement and it is associated with apoptosis and cell cycle arrest.
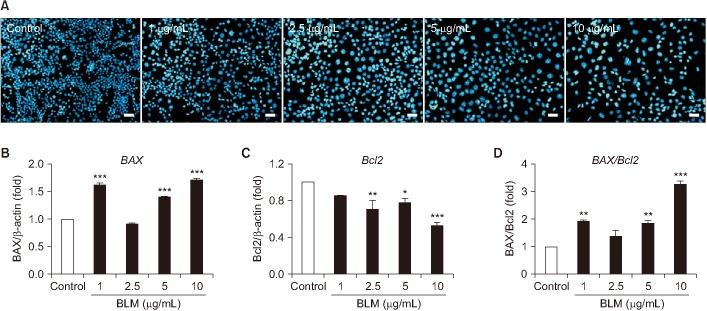 | Figure 2The MLE-12 cells induced apoptosis by bleomycin (BLM). (A) MLE-12 cells were stained with Hoechst 33258 staining after BLM treatment for 24 hours. Scale bars=100 µm. Expression of cell apoptosis marker, BAX (B) and Bcl2 (C) was measured by quantitative real-time reverse transcription-polymerase chain reaction (qRT-PCR). BAX/Bcl2 ratio (D) was divided (B, C) that measured by qRT-PCR. *p<0.05, **p<0.01, ***p<0.001.
|
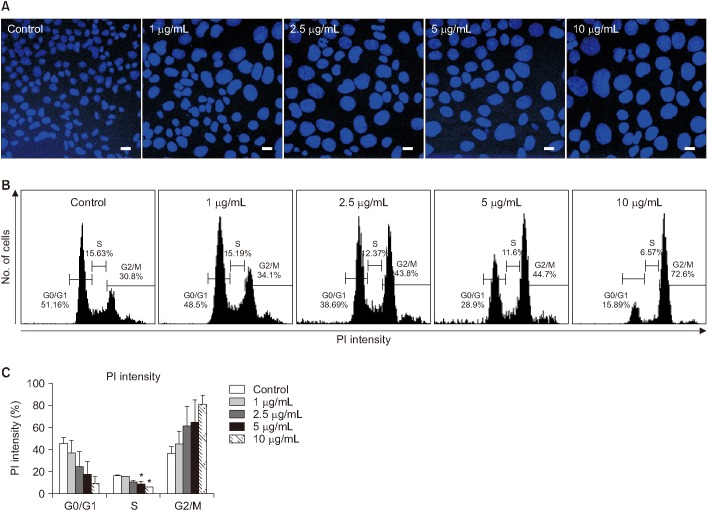 | Figure 3Evaluation of nuclear enlargement and cell cycle arrest induced in MLE-12 cells by bleomycin (BLM). (A) The image was obtained using DAPI staining in MLE-12 cells after BLM treatment. Scale bars=200 µm. (B) Propidium iodide (PI) staining was performed for cell cycle arrest in MLE-12 cells after BLM treatment. (C) Statistical analysis was each percentage of the field in the samples. *p<0.05.
|
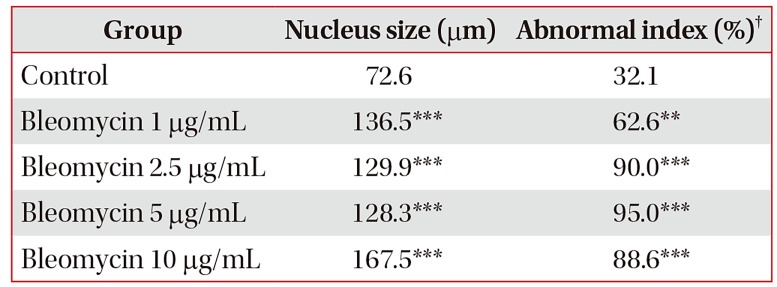
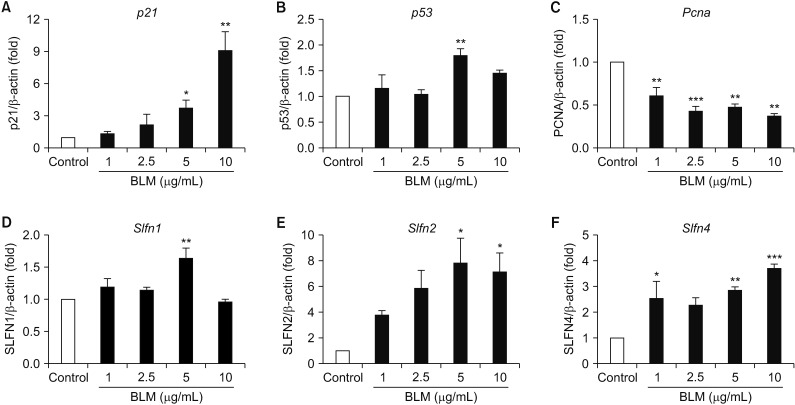
3. BLM-mediated the alteration of cell cycle regulatory gene expression in MLE-12 cells
We next determined the alteration of cell cycle arrest-related gene by BLM treatment for 24 hours in MLE-12 cells. In qRT-PCR analysis, the mRNA levels of p21 and p53 were significantly increased whereas the mRNA level of Pcna was deceased in MLE-12 cells treated with BLM for 24 hours (Figure 4A, B). SLFN family has been implicated the cell cycle regulation and cell growth arrest19. Therefore, we conducted qRT-PCR analysis in BLM-exposed MLE-12 cells, and mRNA levels of SLFN2 and SLFN4 were significantly increased rather than the mRNA expression of Slfn1 (Figure 4D–F). These data suggest that cell cycle arrest-related genes including p21 , p53 , and SLFN family are increased by BLM treatment leading to apoptosis in MLE-12 cells.
 | Figure 4Gene expression associated with cell cycle of bleomycin (BLM) treatment in MLE-12 cells. The MLE-12 cells were treated with BLM (1–10 µg/mL) for 24 hours. Measured the p21 (A), p53 (B), and proliferating cell nuclear antigen (Pcna ) (C) at the mRNA levels, which were associated with cell cycle. BLM exposed MLE-12 cells were measured expression of Slfn1 (D), Slfn2 (E), and Slfn4 (F) by quantitative real-time reverse transcription-polymerase chain reaction analysis.
|
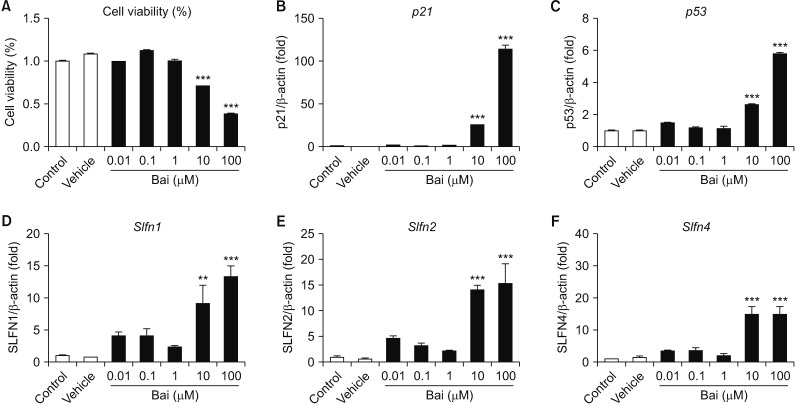
4. Bai-mediated the alteration of SLFN family gene expression in MLE-12 cells
To determine whether cell cycle arrest-related gene and SLFN family gene expression level were changed by Bai that G1/G2 phase cell cycle arrest inhibitor. The cell viability was significantly decreased by Bai in dose-dependent manner (Figure 5A). The mRNA level of cell cycle arrest-related gene such as p21 , p53 , Slfn1 , Slfn2, and Slfn4 were significantly increased by Bai (Figure 5B–F). These data suggest that SLFN1, SLFN2, and SLFN4 associated G1/G2 cell cycle arrest in MLE-12 cells.
 | Figure 5The evaluation of baicalein (Bai) affected MLE-12 cells. The MLE-12 cells were treated with Bai (0.01–100 µM) for 24 hours. (A) The cell viability was measured using MTT assay. Measured the p21 (B) and p53 (C) at the mRNA levels, which were associated with cell cycle. Bai exposed MLE-12 cells were measured expression of Slfn1 (D), Slfn2 (E), and Slfn4 (F) by quantitative real-time reverse transcription-polymerase chain reaction analysis. **p<0.01, ***p<0.001.
|
Go to :
Discussion
Injury of AE II cells has been implicated the pathological feature to trigger the loss of type I alveolar epithelial cells leading to induce the migration of activated fibroblasts in interstitial compartment of distal alveoli in the pathogenesis of pulmonary fibrosis23. Injury of AE II cells includes cellular functional deficiency such as decreased SPC expression which functions to decrease collagen accumulation and inflammation24, increased secretion of pro-inflammatory cytokines25, and accelerated the cellular senescence of alveolar epithelium26. In human A549 cell line and rat primary AE II cells treated with BLM, senescence-associated β-gal activity and flat/enlarged morphology has been exhibited27. In this perspective, the nuclear enlargement and decreased viability of AE II cells in our findings agree with BLM-induced cellular senescence. Moreover, cellular senescence accelerates to undergo cell cycle arrest with distinct morphological feature. Although the cellular senescence in tissue fibrosis is unclear, it is generally accepted that telomeres are shortened in AE II cells of IPF patients28. Recently, Naikawadi et al.29 have been shown that deletion of telomere repeat binding factor 1 (TRF1) which is a component protein in shelterin complex of telomeres of AE II cells causes pulmonary fibrosis in SPC-CreTrf1fl/fl mice. When SPC-CreTrf1fl/fl mice are induced fibrosis, the prolonged telomere dysfunction caused AE II cell hyperplasia during lung remodeling. In contrast, Povedano et al.30 demonstrated that Trf1 deletion develops 90% of AE II cells apoptosis and pulmonary fibrosis through induction of telomere damage using lungs of Trf1Δ/Δ mouse. In addition, Cui et al.31 have been shown that up-regulated miR-34a alveolar epithelial dysfunction is associated with fibrotic lungs and lung myofibroblasts. MiR-34a did not have any obvious difference TGF-β1-induced myofibroblast differentiation; however, the underlied pathway is still conflicting. In this regard, apoptosis is one of roles to be allied in stress-induced morphological change and cellular aging. Accordingly, BLM mediated lung injury and/or pulmonary toxicity is well-known; however, the involved pathway is still remained. Brar et al.32 have shown that oxidative stress causes mitochondrial/nuclear DNA damage and caspase3, poly(ADP-ribose) polymerase, BAX were increased leading to apoptosis in human alveolar epithelial A549 cells. Accordingly, our data agree with BLM-induced apoptosis including up-regulation of pro-apoptotic factor BAX and decreased anti-apoptotic factor Bcl2. Previously, we have shown that cigarette smoke extract similarly induced the apoptotic pattern of apoptosis-related protein family members (Bcl2 and BAX) in mouse lung tissue-derived fibroblasts33. In our data, regulation of Bcl2/Bax was significantly associated with alveolar epithelial cells and pulmonary fibroblasts.
After DNA damage increased p53 and p21 proceeds into a sustained arrest in cell cycle arrest in mammalian cells34. In colorectal cancer cells, the etoposide-induced nuclear enlargement happens strong G2/M phase cell cycle arrest in HCT116 cells35. Additionally, BLM-induced G2 phase cell cycle arrest in the human leukemia-derived cell line36. Our data have shown that BLM treatment induced G2/M phase arrest via nuclear enlargement leading to DNA damage in MLE-12 cells. Recently, Shetty et al investigated that suppression of p53 and miR-34a feedback validates as a potential therapeutic target in a mouse model of BLM, silica, and cigarette smoke exposure and sepsis-induced lung injury37. Aoshiba and colleagues reported that BLM-induced cellular senescence increased the level of p21 in an A549 human lung cancer cell27. Linge et al.38 also have been reported that BLM-induced cell senescence increased the level of p21, p53, and caveolin-1 and occurred G2/M phase arrest. In this study, we have shown that the SLFN family has been included in BLM-mediated apoptosis of alveolar epithelial cells. Despite the diverse biological roles of SLFNs in mammals, it is unknown the associated cellular- and molecular- mechanisms in alveolar epithelial cells. In mouse embryonic fibroblasts, interferon (IFN) induced Slfn genes via STAT1 regulation19. As recent studies suggested, it seems that SLFNs play a critical role in the generation of IFN-inducible responses17. Brady et al.39 have been shown that SLFN1 expression causes cell cycle arrest by inhibiting of mitogen-mediated cycle D1 level in murine CHO epithelial cell line. In our data, when MLE-12 cells were treated with BLM, mRNA levels of Slfn1 , Slfn2 and Slfn4 were significantly increased. In addition, gene expression level of p21, p53, and SLFN1, SLFN2, SLFN4 in MLE-12 cells were increased by Bai. These observations suggest that increased SLFNs expression may associate with cell cycle arrest via regulation of p21/p53 in alveolar epithelial cells. Taken together, we determined that exposure of BLM increases of the release of a pro-inflammatory cytokine, nuclear enlargement, apoptosis and cell cycle arrest in MLE-12 cells. Accordingly, these findings suggest that pulmonary toxicity by BLM may induce DNA damage including SLFN family-related cell cycle arrest. Therefore, we determined whether SLFN family might be involved to understand the pathogenesis of pulmonary fibrosis.
Go to :
 XML Download
XML Download