

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Cryopyrin-associated periodic syndrome (CAPS) is a rare and hereditary autoinflammatory syndrome caused by mutations in NLRP3, which encodes cryopyrin.1 The clinical presentation of CAPS is characterized by periodic systemic inflammation including urticaria, fever and fatigue as well as signs of local inflammation that affects joints, skin, eyes, muscles and the central nervous system. The prevalence of CAPS is estimated at 1 to 3 in 1 million children and adults worldwide2; however, CAPS has not yet been reported in Korea. Establishing a diagnosis of CAPS is challenging because of its rarity; this results in significant delay in diagnosis and treatment. Here, we report 3 cases of CAPS, which comprise the first such reports in Korea, in order to raise awareness of CAPS among Korean clinicians.
This study was approved by the Institutional Review Board of Gangnam Severance Hospital (number: 3-2018-0355). The requirement for informed consent was waived considering the retrospective nature of this study. Verbal informed consent was obtained from all subjects included in this study.
CASE REPORT
Case 1
A 28-year-old man with recurrent urticaria, arthralgia and fever induced by cold stimuli had received symptomatic treatment at other hospitals for 8 years. His father had exhibited the same symptoms since childhood, but did not know the source of these symptoms at the time of his death. This case 1 patient was diagnosed solely with cold urticaria; however, his arthralgia, fever and familial history were not explained by this initial diagnosis. Thus, he visited our hospital for a second opinion.
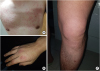
The urticaria developed repeatedly and was worsened by exposure to cold environments. It typically appeared and worsened in the winter; sometimes, it appeared in the summer upon exposure to air conditioning wind. Arthralgia, particularly at the knee joint, accompanied the presentation of urticaria (Fig. 1). Symptomatic treatment, including the use of anti-inflammatory agents, had a minimal effect on the patient's symptoms. He showed elevated levels of C-reactive protein (5.2 mg/dL) and erythrocyte sedimentation rate (62 mm/h); other labs tests were within normal ranges. Antinuclear antibodies were positive, with a titer of 1:80 and a cytoplastic pattern. The patient exhibited a p.Gly303Asp variant of the NLPR3 gene, which is known to cause familial cold autoinflammatory syndrome (FCAS) (Fig. 2).34 He was hesitant to undergo treatment with anti-interleukin (IL)-1 antibody because of the cost.

Case 2
A 2-year-old girl visited our hospital; she had had recurrent urticaria and arthritis (particularly at the knee joint) since birth. She was diagnosed with juvenile rheumatoid arthritis and underwent treatment accordingly; however, recurrent urticaria, fever and arthralgia continued. She exhibited an elevated level of C-reactive protein (9.63 mg/dL). New oral and genital ulcers developed, and a test for HLA B51 was positive. Therefore, she was diagnosed with Behçet's disease and was treated accordingly. However, her symptoms developed again and did not improve. After administration of anakinra, the patient's overall symptoms were much improved and her elevated levels of C-reactive protein returned to normal within 1 month (0.39 mg/dL). Then, genetic testing was conducted under the suspicion of CAPS; finally, a p.Glu569Lys mutation was found in exon 3 of the NLRP3 gene in this patient. Currently, her symptoms are well-controlled with intermittent steroid treatment combined with anakinra.
Case 3
A 4-year-old boy who complained of recurrent urticaria, fever and arthralgia, particularly at the knee joint, was admitted at another hospital, 2 years prior to this case. He was diagnosed with juvenile rheumatic arthritis and was treated with steroid and methotrexate. However, urticaria, fever and arthralgia continued. He was then admitted to our hospital for a second opinion. Genetic testing was conducted under suspicion of CAPS. Before genetic testing reveal the results, tocilizumab was administered to control juvenile rheumatic arthritis and CAPS, both, and all of his symptoms were improved; notably, elevated levels of CRP (10.1 mg/dL) became normalized (0.03 mg/dL). Genetic testing shows finally, a p.Val72Met mutation was found in exon 1 of the NLRP3 gene in this patient. Presently, tocilizumab treatment is continued and the patient's overall symptoms remain well-controlled.
DISCUSSION
Herein, we report 3 cases of CAPS, which comprise the first such reports in Korea. CAPS is a hereditary autosomal-dominant autoinflammatory syndrome caused by mutations in NLRP3, which encodes cryopyrin.1 The clinical manifestation of CAPS is characterized by systemic inflammation, particularly involving fever and fatigue. Local signs affect several tissues such as joints, skin, eyes, muscles and the central nervous system. The most reliable diagnostic model for CAPS suggested by Kuemmerle-Deschner et al.5 is as follows: 1) more than 2 typical symptoms of CAPS (urticaria-like rash, cold-triggered episodes, sensorineural hearing loss, musculoskeletal symptoms, chronic aseptic meningitis and skeletal abnormalities); 2) raised inflammatory markers; and 3) genetic testing. All cases presented in this study met all the above criteria for CAPS. CAPS comprises 3 clinical types including FCAS, Muckle-Wells syndrome (MWS) and chronic infantile neurological, cutaneous, and articular (CINCA) syndrome. FCAS is characterized by cold-induced urticaria and fever. We suspect that the symptoms of the patient in case 1 and his father are concordant with a diagnosis of FCAS. MWS is characterized by systemic amyloidosis and hearing loss. CINCA, a neonatal-onset multisystem inflammatory disease, is characterized by central nervous system inflammation and bone deformities.678 All 3 cases reported here exhibited no evidence of amyloidosis, hearing loss, nervous inflammation, or bone deformities. We suspect that these 3 cases of CAPS can be classified as FCAS; however, long-term follow-up is needed to confirm this diagnosis.
The patients in cases 2 and 3 were treated with monoclonal antibody (anti-IL-1 and anti-IL-6 antibodies, respectively), and symptoms were greatly improved in both cases; however, the patient in case 1 could not be treated with monoclonal antibody because of the cost of treatment. CAPS is caused by a mutation in the NLRP3 gene, which encodes cryopyrin. Cryopyrin plays a central role in innate immunity, as it recognizes intracellular pathogens and some danger signals. In the cytoplasm, cryopyrin interacts with other proteins (caspase recruitment domain inhibitor [CARD] of NF-κB-activating ligands, apoptosis-associated speck-like protein containing a CARD and procaspase 1) to form the canonical inflammasome. This complex activates caspase-1 and eventually cleaves pro-IL-1 into its active form IL-1β.9 This cytokine and downstream inflammatory mediators facilitate systemic and localized responses to infections and injuries. The purpose of treatment for patients with CAPS is to suppress continuous inflammation, prevent damage to organs and other parts of the body, and overall, improve function. Traditionally, cold avoidance and anti-inflammatory drugs have been used as treatment; these approaches can reduce symptoms, but do not change the underlying pathogenesis of excessive IL-1 secretion. Blockage of IL-1 is the primary treatment approach, because IL-1 plays a central role in CAPS pathology. Currently, 3 IL-1 inhibitors, canakinumab, rilonacept and anakinra, are available; the safety and efficiency of these drugs have been reviewed and documented in many studies.101112 However, IL-1 inhibitors have not yet been registered and approved in many countries. Another potent cytokine, IL-6, is generally considered to act downstream of IL-1β.13 Therefore, anti-IL-6 antibodies, such as tocilizumab, are considered for the treatment of CPAS patients.14 It is important to perform quick diagnosis and effective treatment, in order to prevent organ damage.
It is meaningful that this report describes the first CAPS patients in Korea, each of whom was confirmed to exhibit genetic mutations of NLPR3. The father of the patient in Case 1 also exhibited the same symptoms as a child, but did not know the exact etiology of the illness. CAPS itself is extremely rare, and diagnosis of CAPS in Korea may have been further delayed due to the lack of reporting in Korea. In Japan, the clinical characteristics of 19 patients were reviewed in 2012,15 and a study of the long-term safety and efficacy of canakinumab was conducted in 2017.16 Considering the number of patients in Japan nearby, we speculate that there are many cases of CAPS that have not yet been diagnosed in Korea. In previous reports of Korean patients, there was a case in which the CAPS was suspected due to repeated skin rash, arthritis and elevated CRP since childhood; however, the diagnosis of CAPS was not confirmed because a genetic study was not performed.17 Although urticaria and accompanying inflammatory symptoms from childhood are difficult to diagnose with certainty, it is necessary to consider family history and characteristic clinical patterns, in the context of the diagnosis of CAPS and during examination of suspected cases.
By reporting these cases, we hope to increase the awareness and ability to ensure early diagnosis of CAPS. We speculate that it will be helpful to have higher thresholds for clinical doubts regarding CAPS in patients with these symptoms, especially those with family history suggestive of CAPS.
 XML Download
XML Download