

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Inherited retinal diseases (IRD) are characterized by progressive dysfunction of neural retina or retinal pigment epithelium (RPE) which include various subtypes, such as rod-dominant abnormality (rod-cone dystrophy [retinitis pigmentosa, RP]), cone-dominant abnormality (cone or cone-rod dystrophy), macular dystrophy (Stargardt disease, Best macular dystrophy, pattern dystrophy, Sorsby fundus dystrophy, etc.), abnormality of photoreceptors and bipolar cells (X-linked retinoschisis, congenital stationary night blindness [CSNB], etc.), vitreoretinopathies (Wagner syndrome, Stickler syndrome, etc.), and hereditary choroidal diseases.12
Different IRDs may have similar phenotypic characteristics through different genetic mutations. On the other hand, identical genetic mutations may result in IRDs with different phenotypic features.123 Because of the genetic and phenotypic heterogeneity, identification of the causative genetic variants linked to IRD is needed for the accurate diagnosis and treatment of these patients. As the next-generation sequencing (NGS) technology, which can save tremendous cost and time as compared to the previous technologies, has rapidly developed, identification of the causative genes responsible for IRD has dramatically improved.4567891011 To date, about 270 genes have been identified as the cause of IRD in accordance with RetNet database (https://sph.uth.edu/retnet/). The causative genes for IRD are also known to have ethnic and regional differences.5 However, research on this field is mostly based on data obtained from the western population, and no genomic or clinical studies on overall IRD has been previously reported from Korea. In the present study, we have performed molecular analyses involving 86 Korean IRD patients using NGS technology to identify the distribution of the causative genes and mutations linked to IRD in these patients.
METHODS
Participants
We examined 86 Korean patients diagnosed with IRD from unrelated families who visited the Department of Ophthalmology of Seoul National University Bundang Hospital between July 2011 and May 2015 and agreed to participate in the study.
Retinal specialists made the diagnoses of all the IRD cases based on comprehensive ophthalmologic examinations. All the patients underwent visual acuity measurements, slit-lamp biomicroscopy, fundus photography, optical coherence tomography (Spectralis OCT; Heidelberg Engineering, Heidelberg, Germany), and full-field standard electroretinography (VERIS II; Electro-Diagnostic Imaging Inc., San Francisco, CA, USA) according to the protocol of the International Society for Clinical Electrophysiology of Vision.12 Additional examinations such as fundus autofluorescence (Spectralis HRA; Heidelberg Engineering) and electrooculography were performed in selected cases. Pedigrees were constructed based on interviews with the patients. Peripheral blood samples were obtained from all the patients for DNA extraction.
A total of 86 clinically diagnosed IRD patients, including 44 RP, 22 cone dystrophy, 7 Stargardt disease (STGD), 8 macular dystrophy, 1 Best disease, 1 Bardet-Biedl syndrome, 1 CSNB, 1 choroideremia, and 1 occult macular dystrophy were included in the study.
Comprehensive custom gene panel design
We previously reported the development of a custom capture panel of 204 known and candidate genes linked to IRD.13 These genes were selected from RetNet (https://sph.uth.edu/retnet/) and NEIBank (https://neibank.nei.nih.gov/index.shtml), and RetinaCentral (http://neibank.nei.nih.gov/cgi-bin/eyeDiseaseGenes.cgi) (Supplementary Table 1). A total of 204 genes were covered for all coding exons, 5′ and 3′ untranslated regions (UTRs), and each exon flanked alternative splicing areas.
Library preparation and targeted sequencing
The construction of pre-capture libraries (Illumina, Inc., San Diego, CA, USA) and capture process (Roche NimbleGen, Madison, WI, USA) was performed according to the manufacturer's protocols. The captured libraries were sequenced using Illumina HiSeq 2000 using the paired-end (2100 bp) program (Illumina, Inc.).
Bioinformatical analysis
Burrows-Wheeler Aligner was used to align the sequence reads in the human hg19 reference genome. The variants were annotated using GATK packages, SAMtools, and Dindel. The detected variants were annotated using ANNOVAR. NextGENe software (SoftGenetics, State College, PA, USA) was used for the analysis. The 1,000 Genome database, dbSNP137, and the National Heart, Lung and Blood Institute (NHLBI) Exome Sequencing database were used to filter out the common variants. The Human Gene Mutation Database professional database was used to search the known pathogenic mutations. To predict the functional significance of missense variants in programs predicting, amino acid conservation score (SIFT, Polyphen, and MutationTaster) was used. Variant prioritization was performed as described in the previous report.13 Briefly, the variants were selected in case they were not reported in the 1,000 Genome, dbSNP, or NHLBI Exome Sequencing databases or had low frequencies (< 1%) in the Korean population. The variants with less than 30% heterozygous reads or less than 80% homozygous reads were excluded.
The clinical significance of each variant was classified according to the recent recommendations of the American College of Medical Genetics and Genomics (ACMG) on standards for interpretation and reporting of sequence variations: pathogenic, likely pathogenic, uncertain significance, benign, and likely benign variant.14 We primarily used the automated classification by the Intervar and determined the clinical significance via adjustment by manual review.15 All the variants reported in this paper were further confirmed by Sanger sequencing. The disease-causing variants were predicted in consideration with the clinical inheritance pattern and clinical characteristics of each gene.
Ethics statement
The Institutional Review Board (IRB) of Seoul National University Bundang Hospital approved the study protocols (IRB approval No. B-1901-519-103). All patients were fully informed of the purpose and procedures of this study, and written consent was obtained from each participant. All procedures used in this study adhered to the tenets of the Declaration of Helsinki.
RESULTS
The mean coverage of depth per subject was ranged 177–385 folds and 99.6% of the entire region was sequenced at least 10-fold coverage of depth. Overall, the pathogenic mutations in the causative genes were detected in 38/86 (44.2%) of the patients with IRD. Specifically, pathogenic mutations were identified in 18/44 (40.9%) RP, 8/22 (36.4%) cone dystrophy, 6/7 (85.7%) STGD, 2/8 (25%) macular dystrophy, 1/1 (100%) Best disease, 1/1 (100%) Bardet-Biedl syndrome, 1/1 (100%) CSNB, and 1/1 (100%) choroideremia. No causative gene was found for 1 case of occult macular dystrophy (Table 1).
Table 1
Detection rate of causative gene mutations

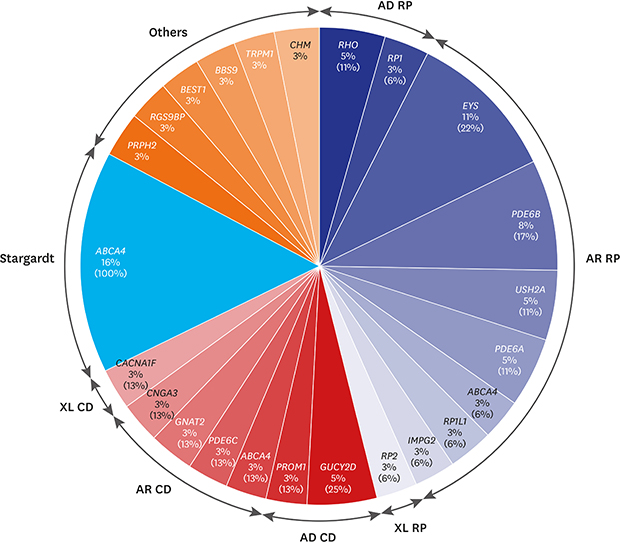
Of the 18 RP cases where the causative genes were identified, pathogenic mutations were detected in 2 cases of RHO, and 1 case of RP1 in 3 autosomal dominant RP (adRP) patients; and 4 cases of EYS, 3 cases of PDE6B, 2 cases of USH2A, PDE6A in each, and 1 case of ABCA4, RP1L1, IMPG2 in each of the 14 autosomal recessive RP (arRP) patients. Mutation in RP2 was detected in 1 case of X-linked RP. All the RP patients presented no signs of systemic symptoms suggesting syndromic RP. Of the 8 patients with cone dystrophy, 2 cases of GUCY2D and 1 case of PROM1 mutations were detected in 3 autosomal dominant cone dystrophy patients. In 4 patients with autosomal recessive cone dystrophy, mutations in ABCA4, PDE6C, GNAT2, and CNGA3 were detected. Mutation in CACNA1F was detected in 1 case of X-linked cone dystrophy. During the follow-up period, all the patients with cone dystrophy had decreased color vision, visual acuity at daytime and decreased photopic response without night-time visual disturbance nor abnormal scotopic response on electroretinography. Although PROM1 p.R373C is a well-known mutation of STGD, the patient with PROM1 p.R373C mutation who was diagnosed as cone dystrophy in this study also showed characteristic features of cone dystrophy. Six cases of autosomal recessive STGD were identified to have mutations in ABCA4. Among 2 patients with unspecified macular dystrophy, 1 case of PRPH2 mutation with autosomal dominant pattern and 1 case of RGS9BP mutation with an autosomal recessive pattern were detected. One case of Best disease, retinitis pigmentosa associated with Bardet-Biedl syndrome, CSNB, and choroideremia had mutations in BEST1, BBS9, TRPM1, and CHM, respectively (Fig. 1 and Table 2). ABCA4 was the most common causative gene in our cohort that was identified in 8 IRD cases including RP (1 case), cone dystrophy (1 case), and STGD (6 cases). Seventeen family samples including 6 patients (2 cone dystrophy, 1 macular dystrophy, 3 Stargardt disease), were additionally analyzed with Sanger sequencing. The result showed that all the 6 patients have compound heterozygous mutations.
Fig. 1
The percentages of patients having the causative genes out of the entire 86 inherited retinal diseases patient cohort are indicated under the gene names and that out of each disease entity such as RP, CD, and Stargardt is indicated in parenthesis.
AD = autosomal dominant, RP = retinitis pigmentosa, AR = autosomal recessive, XL = X-linked, CD = cone dystrophy.
Table 2
Causative genes and mutations in 38 patients with inherited retinal dystrophy

| Diagnosis | Inheritance | Gene | Chromosome | Aminoacid change | Mutation type | CDS | ACMG criteria | Note |
|---|---|---|---|---|---|---|---|---|
| Retinitis pigmentosa (n = 18) | AD | RHO | 3q22.1 | p.T17M | Missense | c.50C>T | Likely pathogenic | Reported28 |
| AD | RHO | 3q22.1 | p.T17M | Missense | c.50C>T | Likely pathogenic | Reported28 | |
| AD | RP1 | 8q11.23-q12.1 | p.Y485X | Nonsense | c.1455T>G | Pathogenic | Reported7 | |
| p.C1399Lfs | Frameshift | c.4196delG | Pathogenic | Reported29 | ||||
| AR | EYS | 6q12 | p.N3123Tfs | Frameshift | c.9368delA | Pathogenic | Novel | |
| p.H2342P | Missense | c.7025A>C | VUS | Novel | ||||
| p.I2188T | Missense | c.6563T>C | Likely pathogenic | Reported30 | ||||
| AR | EYS | 6q12 | p.I2188T | Missense | c.6563T>C | Likely pathogenic | Reported30 | |
| p.G2186E | Missense | c.6557G>A | Likely pathogenic | Reported31 | ||||
| AR | EYS | 6q12 | p.Y841delinsXYfs | Frameshift | c.2522_2523insA | Pathogenic | Reported31 | |
| Splice | Noncoding | c.2382-2A>T | Pathogenic | Novel | ||||
| AR | EYS | 6q12 | p.S1653delinsKXfs | Frameshift | c.4957_4958insA | Pathogenic | Reported31 | |
| AR | PDE6B | 4p16.3 | p.H557Y | Missense | c.1669C>T | Pathogenic | Reported32 | |
| AR | PDE6B | 4p16.3 | p.W290XW | Nonsense | c.869G>A | Pathogenic | Novel | |
| p.A831AV | Missense | c.2492C>T | Likely pathogenic | Novel | ||||
| AR | PDE6B | 4p16.3 | p.S546_I547del | In-frame | c.1636_1641delTCCATC | Likely pathogenic | Novel | |
| p.H557Y | Missense | c.1669C>T | Pathogenic | Reported33 | ||||
| AR | USH2A | 1q41 | p.C934W | Missense | c.2802T>G | Likely pathogenic | Reported34 | |
| p.H68Y | Missense | c.202C>T | VUS | Novel | ||||
| AR | USH2A | 1q41 | p.S1406XS | Nonsense | c.4217C>A | Likely pathogenic | Novel | |
| AR | PDE6A | 5q32 | Splice | Noncoding | c.1407+1G>C | Pathogenic | Reported35 | |
| p.G428GD | Missense | c.1283G>A | Likely pathogenic | Novel | ||||
| AR | PDE6A | 5q32 | Splice | Noncoding | c.1407+1G>C | Pathogenic | Reported35 | |
| p.G428GD | Missense | c.1283G>A | Likely pathogenic | Novel | ||||
| AR | ABCA4 | 1p22.1 | p.Q636K | Missense | c.1906C>A | VUS | Novel | |
| p.Q294X | Nonsense | c.880C>T | Likely pathogenic | Reported8 | ||||
| AR | RP1L1 | 8p23.1 | p.E1559K | Missense | c.4675G>A | VUS | Novel | |
| p.P109delinsSPfs | Frameshift | c.324_325insT | Likely pathogenic | Reported36 | ||||
| AR | IMPG2 | 3q12.3 | Splice | Noncoding | c.828+1G>A | Pathogenic | Novel | |
| XL | RP2 | Xp11.3 | p.C108R | Missense | c.322T>C | Likely pathogenic | Novel | |
| Cone dystrophy (n = 8) | AD | GUCY2D | 17p13.1 | p.F883Lfs | Frameshift | c.2649delT | Likely pathogenic | Reported37 |
| p.R964L | Missense | c.2891G>T | VUS | Novel | ||||
| AD | GUCY2D | 17p13.1 | p.R838H | Missense | c.2513G>A | Likely pathogenic | Reported38 | |
| AD | PROM1 | 4p15.32 | p.R373C | Missense | c.1117C>T | Pathogenic | Reported39 | |
| AR | ABCA4 | 1p22.1 | p.L1583P | Missense | c.4748T>C | VUS | Reported8 | |
| p.Q636K | Missense | c.1906C>A | VUS | Novel | ||||
| AR | PDE6C | 10q23.33 | p.W548L | Missense | c.1643G>T | VUS | Novel | |
| p.G836Efs | Frameshift | c.2507delG | Likely pathogenic | Novel | ||||
| AR | GNAT2 | 1p13.3 | p.H244Sfs | Frameshift | c.730_743delCATGAGTCTTTGCA | Likely pathogenic | Novel | |
| p.R161X | Nonsense | c.481C>T | Likely pathogenic | Reported40 | ||||
| AR | CNGA3 | 2q11.2 | p.L185V | Missense | c.553C>G | VUS | Reporteda | |
| p.R283Q | Missense | c.848G>A | Pathogenic | Reported41 | ||||
| XL | CACNA1F | Xp11.23 | p.G1350D | Missense | c.4049G>A | Likely pathogenic | Novel | |
| Stargardt disease (n = 6) | AR | ABCA4 | 1p22.1 | p.R2049fs | Frameshift | c.6146delA | Likely pathogenic | Reported8 |
| p.D654N | Missense | c.1933G>A | VUS | Reported8 | ||||
| AR | ABCA4 | 1p22.1 | p.R2049fs | Frameshift | c.6146delA | Likely pathogenic | Reported8 | |
| p.T1117A | Missense | c.3349A>G | Likely pathogenic | Novel | ||||
| AR | ABCA4 | 1p22.1 | p.C1140W | Missense | c.3420C>G | Likely pathogenic | Reported42 | |
| p.I1114_M1115delinsM | In-Frame | c.3342_3344delCAT | Likely pathogenic | Novel | ||||
| AR | ABCA4 | 1p22.1 | p.C1140W | Missense | c.3420C>G | Likely pathogenic | Reported42 | |
| p.I1114_M1115delinsM | In-Frame | c.3342_3344delCAT | Likely pathogenic | Novel | ||||
| AR | ABCA4 | 1p22.1 | p.L1157X | Nonsense | c.3470T>G | Likely pathogenic | Reported8 | |
| p.R290Q | Missense | c.869G>A | VUS | Novel | ||||
| AR | ABCA4 | 1p22.1 | p.N1588Y | Missense | c.4762A>T | VUS | Novel | |
| p.L1157X | Nonsense | c.3470T>G | Likely pathogenic | Reported8 | ||||
| Macular dystrophy (n = 2) | AD | PRPH2 | 6p21.1 | p.P219_P221delinsP | In-Frame | c.657_662delACGGCC | Likely pathogenic | Novel |
| AR | RGS9BP | 19q13.11 | p.E71X | Nonsense | c.211G>T | Likely pathogenic | Reported43 | |
| p.G205delinsGLfs | Frameshift | c.614_615insG | Pathogenic | Novel | ||||
| Best disease (n = 1) | AD | BEST1 | 11q12.3 | p.N296K | Missense | c.888C>A | Likely pathogenic | Reporteda |
| Syndromic RP associated with BBS (n = 1) | AR | BBS9 | 7p14.3 | p.V260G | Missense | c.779T>G | VUS | Novel |
| p.Q807X | Nonsense | c.2419C>T | Likely pathogenic | Novel | ||||
| CSNB (n = 1) | AR | TRPM1 | 15q13.3 | p.N1304Ifs | Frameshift | c.3911delA | Pathogenic | Novel |
| p.R1133XR | Nonsense | c.3397C>T | Pathogenic | Novel | ||||
| Choroideremia (n = 1) | XL | CHM | Xq21.2 | p.Q63X | Nonsense | c.187C>T | Pathogenic | Novel |
DISCUSSION
In the present study, targeted exome sequencing (gene panel) was employed for the genetic diagnosis of Korean patients with diverse IRD. Molecular diagnosis was possible in 38 (44.2%) cases. To the best of our knowledge, this is the first study to evaluate the diagnostic rate and causative genes responsible for causing diverse IRD among Korean patients using a gene panel approach.
Over the past decades, IRD has been regarded as an ideal target for gene therapy for its monogenic trait, direct accessibility of the affected cells by various surgical procedures, identification of many causative genes, and immune-privileged environment of the retina.3 Recently, the Food and Drug Administration has approved voretigene neparvovec-rzyl (Luxturna®, Spark Therapeutics, Inc., Philadelphia, PA, USA), the first gene therapy using adeno-associated virus as the vector carrying (targeting) RPE65, which is responsible for causing Leber congenital amaurosis and early onset RP.16 Furthermore, numerous clinical trials targeting IRD-causing genes including CNGB3, CNGA3, REP1, CHM, ND4, RS1, MERTK, RPGR, and ABCA4 are in progress.317 To use this genetic treatment modality, identification of the causative genes must precede before establishing a concrete treatment strategy. Considering that the distribution of the causative genes varies among ethnicities, it is necessary to identify the causative genes in various forms of IRD patients in Korea for the upcoming era of gene therapy.
Because RP is the most common form of IRD, genetic studies of IRD have mainly focused on the different mutations in RP.18 To date, 71 causative genes and loci have been identified to be linked to RP (RetNet [http://www.sph.uth.tmc.edu/RetNet]). In a previous study involving parallel sequencing of 53 targeted RP genes in 62 non-syndromic Korean patients, casual variants were detected in 50% of the patients.7 In that study, PRPF31 (30%), RHO (20%), and RP1 (20%) were the frequently affected genes in adRP, whereas EYS (40%) and PDE6B (40%) were the frequently affected genes in arRP. We currently identified 18/44 (40.9%) causative gene mutations in the overall RP patients, including RHO (67%) in adRP and EYS (29%) in arRP. Our results are consistent with other recent studies conducted in east Asia demonstrating a higher incidence of EYS mutation in arRP than in the studies conducted among the Caucasians.7919202122 In terms of X-linked RP, RP2 variant was identified uniquely in both the Korean studies, including ours, instead of RPGR which is thought to be the most common causative gene for X-linked RP. The reason for this low detection rate of RPGR in the Korean studies might be due to the ethnic difference or the repetitive purine-rich ORF15 region, a mutation hotspot in RPGR, which is poorly covered using NGS.7
Mutations in five genes (ELOVL4, PROM1, PRPH2, BEST1, and ABCA4) have been reported to be responsible for STGD or Stargardt-like disease. About 47%−96% of the patients with clinically diagnosed STGD were reported to have pathogenic mutations identified, and most of the cases showed autosomal recessive ABCA4 mutations.4102324 The reason for a relatively higher detection rate of the causative genes in STGD than in the other IRDs might be due to the characteristic distinctive features of STGD−choroidal silence sign in fundus fluorescein angiography. This implies that the fine distinction among the different phenotypes is important for the differential diagnosis of IRD. Especially in Asia, the causative genes of STGD other than ABCA4 are rarely found.232526 In a recent study that included clinically diagnosed Korean STGD patients, 17 out of 24 (70.8%) patients were detected to have ABCA4 mutations.8 No potential mutations in the ELOVL4 and PROM1 were, however, found. Similarly, we have detected ABCA4 mutations in 6 of 7 (85.7%) Korean STGD patients and no other 4 STGD-associated genes were found.
Mutations in the KCNV2, CNGB3, and ABCA4 were reported to be the common causative factors of autosomal recessive cone dystrophy, whereas, GUCY2D, CRX, and GUCA1A were accounted for more than half of the genetically identified cases of autosomal dominant cone dystrophy.127 Till date, there is no report of a genetic study with cone dystrophy in Korea. We have identified the causative genes in 8 (36.4%) out of 22 Korean cone dystrophy patients with a high rate of detection of the known causal genes.
The current study could not identify any possible causative genes in about 56% of the cases. Several reasons could explain this low detection rate. First, uninvolved genes or non-exon sequences in our diagnostic panel or unknown causative genes for IRD may exist. Second, ethnic differences, as mentioned, might contribute to the presence of undiscovered Asia-specific causative genes and pathogenic variants linked to IRD. In particular, there is a possibility that specific causative genes and pathogenic variants for IRD exists in Korean population. Third, genomic structural variants and genomic rearrangements affecting more than 50 bp are not detectable by whole exome sequencing or gene panel, unlike whole genome sequencing. The hospital-based retrospective design is also a limitation of the study. Our data might have been affected by selection bias, and hence, might not reflect the true prevalence and genetic association in IRD in the Korean population. However, we have enrolled consecutive IRD patients who were referred from the local clinics without imposing any exclusion criteria. Thus, we have collected crude data on the prevalence and distribution of genetic mutations among the IRD patients in the Korean population. Further population-based studies are necessary to reveal the detailed genetic epidemiology of IRD in the Korean population.
In conclusion, to date, the present study has screened the largest sample of Korean patients diagnosed with different types of IRD and described the relevant genetic characteristics in the cohort. Considering that different subtypes of IRD are genetically and phenotypically heterogeneous and the causative genes of IRD have ethnic differences, our data will serve as a basis for understanding the genetic distribution and characteristics of the Korean IRD patients.
 XML Download
XML Download