

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Cardiovascular complications are the main cause that determines mortality in patients suffering from diabetes, mainly due to increased ischemic heart disease in diabetics. Of note though, an increased risk for the development of heart failure remains after adjusting for concomitant risk factors such as coronary artery disease (CAD) and hypertension.1 Thus, the term diabetic cardiomyopathy (DC) was introduced, defined as ventricular dysfunction in the absence of CAD and hypertension. Today, DC is increasingly recognized as an independent cardiac entity in clinical practice. Cardiac hypertrophy and diastolic dysfunction are typical clinical features of DC, which may progress to heart failure with preserved ejection fraction (HFpEF).234 Although not readily detectable using conventional echocardiography, subtle abnormalities in systolic function may be detected using strain analysis and measurements of peak systolic velocity.5 Recently, Seferovic and Paulus3 even called DC a “two-faced disease” that may not only be characterized by a restrictive phenotype leading to HFpEF but also a dilated phenotype leading to heart failure with reduced ejection fraction (HFrEF). While the development of a meaningful systolic dysfunction in DC remains subject of debate, many studies performed in diabetic animals and humans provided strong data of structural and cellular alterations that should adversely impact the response of the heart to subsequent stressors, and that partially overlap with derangements observed in failing hearts of other etiologies. The number of mechanisms proposed to cause DC steadily grows and adds to the complexity of this cardiac entity. This review aims to provide an overview and update of established underlying mechanisms of DC, as well as a discussion of recently identified and emerging mechanisms that may also contribute to the structural and functional alterations in DC.
ESTABLISHED MECHANISMS OF DC
1. Myocardial fatty acid (FA) metabolism and lipotoxicity in DC
The myocardial metabolic phenotype in diabetes is characterized by increased FA uptake and oxidation combined with a decrease in glucose oxidation. This substrate oxidative pattern is mainly mediated by competition between glucose and FA metabolism as defined by the Randle hypothesis and by increased activation of peroxisome proliferator-activated receptor α (PPARα) signaling, which increases the expression of proteins and enzymes involved in FA uptake and oxidation.6 Indeed, overexpression of PPARα results in a cardiac phenotype that resembles molecular alterations observed in animal models of DC, including repression of glucose utilization, cardiac hypertrophy and systolic dysfunction.7 Increased FA oxidation may induce mitochondrial uncoupling by reactive oxygen species (ROS)-mediated direct activation without an increase in levels of mitochondrial uncoupling proteins, which may result in decreased ATP regeneration and an impairment in cardiac efficiency (cardiac work/oxygen consumption).89 The shift towards increased FA utilization even remains consistent after application of insulin in type 2 diabetes, which indicates a metabolic inflexibility of the diabetic heart to easily switch between FA and glucose utilization.10 As a consequence, the adaptation to a chronic increase in energy demand (e.g. hypertension) or myocardial ischemia reperfusion, which both may require an increase in glucose utilization to maintain cardiac structure and function, may be impaired in the diabetic heart.1112
Since FA uptake exceeds the FA oxidative capacity of mitochondria in DC, non-oxidized FAs may be diverted into other pathways of lipid metabolism, including the synthesis of triacylglycerols (TAG), ceramides, diacylglycerols, long-chain acyl-CoAs, and acyl-carnitines.13 One mechanism contributing to the lipotoxic component of DC may represent a dysbalance between FA uptake and oxidation mediated by activation of glycogen synthase kinase-3α (GSK-3α) during high fat diet.14 Activation of GSK-3α may lead to a specific phosphorylation of PPARα, thereby inducing uptake and storage of FAs but not oxidation of FAs and thus leading to an intensified lipid accumulation.14 Knock out of GSK-3α attenuates cardiomyocyte hypertrophy and the development of diastolic dysfunction.14 Increased TAG storage is widely considered a biomarker of excess lipid storage in DC, but may actually represent an inert buffer system to divert FA away from pathways that generate toxic lipid intermediates, although some studies also suggest that TAG accumulation may contribute to cardiac hypertrophy and dysfunction.1315 Good evidence exists though that ceramide accumulation may be a culprit lipid intermediate mediating toxic effects in DC. Increased myocardial ceramide levels are associated with cardiac dysfunction in rodent models of diabetes, and pharmacologic inhibition of ceramide biosynthesis improved cardiac function and attenuated cardiomyocyte apoptosis in lipotoxic cardiomyopathy and diabetic Zucker diabetic fatty rats, respectively.1617 Interestingly, a milk fat-based high fat diet rich in myristate caused cardiac hypertrophy and dysfunction which was induced by ceramide synthase 5-mediated production of C14-ceramide, and ceramide-induced cardiac hypertrophy may have been caused by increased autophagy in this model, suggesting that maybe not bulk but specific ceramide species may have deleterious lipotoxic effects in DC.18
2. Glucose utilization and toxicity in DC
Hyperglycemia causes a glucose gradient across the cell membrane since serum glucose availability is high but the intracellular capacity to oxidize glucose is rather low due to impaired pyruvate dehydrogenase (PDH) activity.19 As a consequence, glucose uptake is increased by mass action effects, resulting in accumulation of glycolysis intermediates which may enter additional pathways of glucose utilization that may exert harmful effects on the heart in diabetes.20 Accumulation of glucose-6-phosphate results in increased generation of nicotinamide adenine dinucleotide phosphate (NADPH) by the activity of glucose-6-phosphate dehydrogenase and subsequent enzymatic steps of the pentose phosphate pathway, thereby providing a substrate for cytosolic ROS production by NADPH oxidases (NOX).21 Glucose-6-phosphate can also contribute to the formation of harmful advanced glycation end products (AGEs).22 AGE result from glycation of free amino groups of predominantly long-lived proteins and may harm cardiomyocytes through direct formation of cross-linked macro-molecules. AGE may impair Ca2+ handling by inducing dysfunction of sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA) 2a, may increase fibrosis by inducing cross-linking of collagen molecules, and may bind to the AGE receptor (RAGE) and thereby increase nuclear factor (NF)-κB signaling to increase collagen production and promote proinflammatory signaling in DC.23242526 Finally, fructose-6-phosphate may enter the hexosamine pathway, which may lead to increased O-GlcNAcylation of proteins and thus impaired Ca2+ handling in DC, among other effects (see also 3.4).27 The alterations of cardiomyocyte glucose and FA metabolism and their potential consequences in DC are schematically illustrated in Fig. 1.
Fig. 1
Consequences of diabetes-induced alterations in substrate metabolism and sources of increased ROS in DC. Increased FA uptake results in accumulation of TAG, lipid intermediates and increased FA oxidation, all of which may harm the cardiomyocyte. Increased glucose entry into the cardiomyocyte may promote alternative pathways of glucose utilization, thus provoking an increase in ROS, AGE, and O-GlcNAcylation. Increased amounts of intramitochondrial ROS may be generated by the electron transport chain, by increased MAO activity or by increased calpain-1 levels, whereas NOX2, NOX4, and XO may contribute to increased cytosolic ROS generation.
ROS, reactive oxygen species; DC, diabetic cardiomyopathy; FA, fatty acid; TAG, triacylglycerols; AGE, advanced glycation end product; NOX, NADPH oxidases; XO, xanthine oxidase; GLUT, glucose transport protein; CD, cluster of differentiation; FATP, fatty acid transport protein; ACS, acetyl-CoA synthetase; PPARα, peroxisome proliferator-activated receptor α; FAO, fatty acid oxidation; PPP, pentose phosphate pathway; NADPH, nicotinamide adenine dinucleotide phosphate; HBP, hexosamine biosynthetic pathway; O-GlcNAc, O-linked β-N-acetylglucosamine; SERCA, sarco(endo) plasmic reticulum Ca2+-ATPase; DAG, diacylglycerols; TAG, triacylglycerols; CPT, carnitine palmitoyltransferase; TCA, trichloroacetic acid; MAO, monoamine oxidases; PDH, pyruvate dehydrogenase; ADP, adenosine diphosphate; ATP, adenosine triphosphate; UCP, uncoupling protein.

3. Oxidative stress in DC
Oxidative stress is widely accepted to contribute to the pathogenesis of DC (Fig. 1), as evidenced by increased mitochondrial H2O2 emission, increased levels of peroxidation products, increased protein tyrosine nitration, and induction of the antioxidant defense system in diabetic animals or humans.8282930 A causative contribution of mitochondrial oxidative stress was emphasized by studies investigating the effects of ROS scavenging on DC using transgenic overexpression of catalase, manganese superoxide dismutase or peroxiredoxin-3 in diabetic mice, which at least partially restored mitochondrial function, attenuated apoptosis, improved cardiomyocyte contractility, and attenuated ROS-induced NF-κB-mediated cardiac inflammation.313233 Similarly, treatment with the mitochondria-targeted antioxidant mito-TEMPO attenuated mitochondrial oxidative stress, apoptosis, cardiac hypertrophy and cardiac dysfunction in diabetic mice.34 Beyond mitochondria, superoxide generation by NOX also contributes to oxidative stress in the diabetic heart.35 Activity or expression of NOX2, NOX4 as well as adaptor proteins of the NADPH oxidase complexes are increased in diabetic hearts, and treatment of diabetic mice with the NOX inhibitor apocynin attenuated increased superoxide generation and improved cardiac dysfunction.3637 Xanthine oxidase is another extramitochondrial ROS-generating enzyme whose activity is increased in diabetic hearts.38 Inhibition of xanthine oxidase by allopurinol treatment decreases oxidative stress, fibrosis, and cardiac dysfunction in diabetic rodents.3839 Finally, a decrease in the dimer to monomer ratio of endothelial NO synthase (eNOS) may contribute to oxidative stress in diabetic hearts.40 Monomerization of eNOS, i.e. uncoupling of eNOS, results in an increased production of ROS instead of NO, and eNOS uncoupling has been observed in diabetic hearts.41 Importantly, inhibition of NOS uncoupling by sepiapterin attenuated cardiac dysfunction in diabetic mice 41. In addition, superoxide reacts with NO to form peroxynitrite, a very reactive oxidant. Peroxynitrite promotes apoptosis, activates mitogen-activated protein kinases (MAPKs) and poly(ADP-ribose)-polymerase 1 (PARP-1), impairs intracellular Ca2+ handling, and impairs cardiac contractility.42
4. Fibrosis in DC
Interstitial and perivascular deposition of collagen is typically observed in DC both in animal models and human subjects and may contribute to diastolic and systolic dysfunction.4344 Interstitial fibrosis increases stiffness of the left ventricle and thereby contributes to impaired relaxation and diastolic dysfunction. Fibrosis may also impair the transduction of cardiomyocyte contraction into myocardial force development, leading to uncoordinated contraction.45 Collagen and other extracellular matrix proteins are produced by fibroblasts, whereas fibrogenic mediators and signaling molecules are secreted by inflammatory cells (e.g. macrophages) and cardiomyocytes.46 In DC, increased collagen production may be driven by increased expression of signaling receptors that regulate collagen expression such as transforming growth factor β (TGFβ), or transcription factors such as connective tissue growth factor (CTGF), or may be related to increased activation of the DNA repair enzyme PARP-1.474849 Opposite to increased collagen deposition, dysregulation of extracellular matrix degradation due to remodeling of matrix metalloproteinases, in particular reduced expression of matrix metalloproteinase 2, may also contribute to accumulation of increased connective tissue content in diabetic hearts.50
5. Forkhead box transcription factors (FoxO) in DC
FoxO are a family of transcriptions factors that contain a specific amino acid sequence called forkhead box which they use to bind to target DNA sequences. In general, FoxO signaling regulates the expression of many genes, including genes encoding for proteins involved in cellular metabolism, oxidative stress, apoptosis and cell cycle differentiation. FoxO1, FoxO3 and FoxO4 are expressed in the heart. While deletion of FoxO3 or FoxO4 does not affect cardiac development, deletion of FoxO1 results in embryonic lethality by day E10.5–E11 and severe defects in vascular and cardiac growth, implying a predominant role of the FoxO1 isoform for cardiac physiology.51 Myocardial expression of FoxO1 is increased in models of type 2 diabetes, and various stimuli like hyperglycemia, lipid overload, ROS, cytokines and other growth factors have been shown to regulate FoxO1 activity in DC.5152 Importantly, mice lacking FoxO1 are protected from high-fat diet-induced impairment in glucose uptake, cardiomyocyte insulin resistance, cardiomyocyte hypertrophy, lipid accumulation and cardiac dysfunction, strongly implicating increased activity of FoxO1 in the pathogenesis of DC.52
6. Inflammation in DC
Diabetes is a proinflammatory state and chronic low-grade inflammation in the heart may contribute to the pathogenesis of DC.5354 A number of studies reported increased activation of the proinflammatory transcription factor NF-κB, increased expression of inflammatory cytokines (interleukin [IL]-1β, IL-6, IL-18, tumor necrosis factor [TNF] α, TGFβ-1), increased expression of cell adhesion molecules (ICAM-1, VCAM-1), and increased infiltration of macrophages and leukocytes in DC.505556 Proinflammatory signaling may contribute to cardiac dysfunction by increasing oxidative stress and peroxynitrite levels, which may cause direct cellular damage, may increase apoptotic cell death and may impair intracellular Ca2+ handling, for example by impairing expression and activity of SERCA2a.57 Myocardial inflammation may also increase fibrosis, thereby contributing to diastolic and systolic dysfunction.58 Accordingly, a number of studies demonstrated attenuation of contractile dysfunction in DC by decreasing oxidative stress, fibrosis and apoptosis by various different interventions that reduced cardiac inflammation, including AT-1 receptor antagonism, activation of the kallikrein-kinin system, inhibition of p38 MAPK signaling, gene deletion of kinin receptor b1, inhibition of interleukin converting enzyme, anti-TNFα treatment, inactivation of GSK-3β, and cannabidiol treatment.38505556 Furthermore, myocardial expression levels of the pattern recognition receptor toll-like receptor 4 (TLR4), a key proximal signaling receptor responsible for initiating the innate immune response, is increased in DC, and TLR4 silencing prevents cardiac lipid accumulation, myocardial apoptosis, ventricular remodeling and dysfunction, and suppresses ROS production in diabetic hearts.596061 Yet another inflammatory mechanism contributing to DC may be increased myocardial NLR family pyrin domain containing 3 (NLRP3) inflammasome activation, a multi protein complex which may induce apoptosis via activation of caspase-1, and which may induce an inflammatory form of programmed cell death termed pyroptosis (characterized by cytoplasmic swelling, plasma membrane rupture and nuclear DNA damage).62 Expression of NLRP3, swelling of mitochondria and fibrils, and caspase-1 dependent pyroptosis are increased in diabetic hearts.3363
7. Apoptosis in DC
Apoptosis is a highly controlled mechanism of programmed cell death and seems to be the dominant form of cell death in DC, compared to lower rates caused by necrosis.536465 Due to the poor ability of the heart to regenerate cardiomyocytes increased apoptotic cell death is considered to contribute to contractile dysfunction in DC.66 Increased apoptosis was observed in right atrial appendage of diabetic patients, which was partially inhibited either by inhibition of PARP-1 or by inhibition of caspase-3, the common downstream effector of extrinsic and intrinsic activation of apoptotic signaling.65 In type 1 diabetic animals, both increased death receptor signaling and mitochondria-dependent pro-apoptotic signaling (increased caspase-9 activity, increased Bak/Bax expression, mitochondrial cytochrome c release) contribute to increased apoptosis in DC, and antioxidant treatment attenuated apoptosis and both of these signaling pathways, suggesting a significant role of increased ROS in apoptosis induction in DC.6467 A recent study also suggests that dissociation of Bcl-2 from beclin-1 by restoration of impaired AMP-dependent protein kinase (AMPK) activity may attenuate apoptosis in DC by restoring autophagy, supporting the proposal that an interplay between autophagy and apoptosis may be important in DC.68 Furthermore, endoplasmic reticulum stress may promote apoptosis in DC by activating JNK signaling and apoptosis via the intrinsic and extrinsic pathway, or by increasing PERK/CHOP signaling, which may trigger apoptosis by switching expression towards pro-apoptotic Bcl-2 proteins.69
8. Impaired Ca2+ handling in DC
During each contraction cycle, cardiomyocyte action potentials trigger Ca2+ entry via L-type Ca2+ channels (LTCC), which triggers sarcoplasmic reticulum (SR) Ca2+ release via ryanodine receptors (RyR), resulting in increased cytosolic Ca2+ concentration, binding to troponin C and triggering of actin myosin interaction, i.e. cardiac contraction. Relaxation occurs mainly by Ca2+ reimport into the SR by SERCA2a and by Ca2+ export via Na+/Ca2+ exchanger (NCX). In diabetes, Ca2+ entry, intracellular Ca2+ cycling, and Ca2+ efflux are altered both in animal models and humans, contributing to impaired cardiac contraction and relaxation (Fig. 2). Reduced Ca2+ entry is the consequence of both reduced expression and altered voltage dependence of LTCC.70 Impaired intracellular Ca2+ cycling includes decreases of the amplitude of Ca2+ and of the systolic rate of Ca2+ rise and decay.2425 Prolonged rates of Ca2+ decay may result from impaired SERCA2a activity during diastole, which may lead to a reduction in SR Ca2+ storage of up to 50% and thus contributes to impaired relaxation and diastolic dysfunction.71 Impaired Ca2+ efflux may be mainly related to reduced expression and activity of NCX.24 In addition, mitochondrial Ca2+ uptake and release which follows the cytosolic changes in Ca2+ content are impaired in DC, which may compromise the activity of Ca2+-sensitive TCA cycle enzymes and of the F0F1-ATPase and thus of oxidative ATP regeneration.7273 In models of type 2 diabetes, contractile dysfunction may be driven by a significant reduction of the Ca2+ transient due to decreased Ca2+ influx as a consequence of decreased LTCC expression, by decreased SR Ca2+ content due to reduced SERCA2a expression and increased phospholamban expression, and by decreased content and activity of RyR.7475 In addition, hyperglycemia may cause O-GlcNAcylation of Ca2+/calmodulin-dependent protein kinase II (CaMKII), which may increase diastolic SR Ca2+ leak via RyRs leading to SR Ca2+ depletion.76
Fig. 2
Potential mechanisms and effects of impaired Ca2+ handling in DC. Impairment in cardiomyocyte Ca2+ influx and efflux, impaired release and reuptake of Ca2+ by the SR, and impaired Ca2+ uptake by the mitochondria which may subsequently impair ATP regeneration and thereby contribute to systolic and diastolic dysfunction in DC.
DC, diabetic cardiomyopathy; SR, sarcoplasmic reticulum; ATP, adenosine triphosphate; NCX, Na+/Ca2+ exchanger; LTCC, L-type Ca2+ channels; TCA, trichloroacetic acid; DH, dehydrogenase; RyR, ryanodine receptor; CaMKII, calmodulin-dependent protein kinase II; O-GlcNAc, O-linked β-N-acetylglucosamine; SERCA, sarco(endo)plasmic reticulum Ca2+-ATPase; AGE, advanced glycation end product.
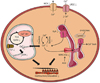
9. Renin-angiotensin-aldosterone system (RAAS)
Diabetes and hyperinsulinemia induce both systemic activation and local myocardial activation of the RAAS, thereby promoting fibrosis, apoptosis, and oxidative stress.777879 Antagonizing myocardial aldosterone action or inhibition of angiotensin converting enzyme reduces oxidative stress, apoptosis and fibrosis, thereby attenuating cardiac hypertrophy and improving myocardial function in DC, suggesting a strong contribution of RAAS activation in the pathogenesis of DC.506779808182 Mechanisms underlying these beneficial effects of RAAS antagonism in DC include a decrease in MMP-2 activity, a decrease in TGFβ expression, inhibition of MAPK signaling such as p38 MAPK, and inhibition of NADPH oxidases, among others.506177
EMERGING MECHANISMS OF DC
1. Autophagy in DC
Autophagy is a highly regulated mechanism of programmed cell death that is indispensable for prenatal and postnatal development, maturation and function of the heart.8384 Damaged cellular components (proteins, lipids, mitochondria, etc.) are engulfed in double membrane structures called autophagosomes, which fuse with lysosomes to break down the sequestered material by hydrolysis. The entire process of autophagy-mediated removal of cellular components includes more than 30 autophagy-related genes (Atg) and can be evaluated by measuring markers of autophagic activity such as LC3-II, p62, or Beclin-1. A low level of constitutive autophagy seems to be protective for a healthy heart.83 In DC, the role of myocardial autophagy may differ between type 1 and type 2 diabetes. In type 1 diabetic OVE26 mice, decreased protein levels of LC3-II, Beclin-1, and a decreased number of autophagosomes was associated with impaired AMPK activity and cardiac dysfunction, and activation of AMPK by metformin treatment restored autophagy and cardiac dysfunction.85 Similarly, reduced expression of LC3-II and Beclin-1 was associated with impaired AMPK activity and cardiac dysfunction in streptozotocin (STZ)-diabetic mice, and overexpression of heme oxygenase-1 restored AMPK activity, autophagy and cardiac function.86 Thus, it has been proposed that a suppression of autophagy in models of type 1 diabetes may result from impaired AMPK activity and may contribute to cardiac dysfunction. Of interest though, cardiac dysfunction and damage in STZ-diabetic and OVE26 mice was attenuated by heterozygous deletion of Beclin-1 but was exacerbated by overexpression of Beclin-1, thus suggesting that diminished autophagy could also be an adaptive mechanism that limits cardiac dysfunction in DC of type 1 diabetes.87 It also remains to be determined whether the beneficial effects of restoration of AMPK activity result from restoration of autophagy or from improvement of impaired myocardial energetics by activation of oxidative ATP regeneration. More studies are needed to clarify the role of autophagy in DC of type 1 diabetes.
In models of insulin resistance and type 2 diabetes, autophagy was mostly reported to be suppressed in response to high fat feeding, although activation of autophagy was reported in studies using a milk-fat based diet or high fructose feeding.88 Supporting a maladaptive role of autophagy suppression in type 2 diabetes models, deletion of Akt2 enhanced cardiac autophagic flux and reduced cardiac hypertrophy and cardiac dysfunction in mice undergoing high fat feeding, and treatment with the mTOR inhibitor rapamycin improved cardiac remodeling and dysfunction in high fat fed adiponectin knockout mice.8990 In contrast, induction of autophagy was required for cardiac hypertrophy and dysfunction in response to milk-fat feeding, suggesting that increased autophagy may contribute to cardiac pathology in DC of type 2 diabetes.18 Some of the controversial results both in type 1 and type 2 diabetes may be related to differences in the systemic diabetic milieu among the animal models. Indeed, high glucose incubation is capable of inhibiting autophagy whereas palmitate is able to induce autophagy in cardiac cells, and serum levels of both metabolites are increased in animal models of diabetes.9192 Furthermore, both cardiomyocyte insulin resistance and insulin deficiency, which occur to varying degrees in the distinct models of diabetes, will impair cardiomyocyte insulin action, and impaired cardiomyocyte insulin signaling due to loss of IRS-1 and IRS-2 has been shown to result in unrestrained autophagy, leading to early postnatal development of heart failure.84 While emerging evidence is highly suggestive of a significant role of altered autophagy in DC, elucidating the adaptive or maladaptive nature of suppressed and/or increased autophagy in DC will be one of the challenges of future studies.
2. Mitochondrial dynamics and quality control in DC
Mitochondrial dynamics determine mitochondrial size and morphology and are important for proper maintenance of mitochondrial membrane potential, electron transport capacity, and ROS production in the heart. Both deletion of mitochondrial fusion proteins (Opa1, Mfn1/Mfn2) or fission proteins (Drp1) may result in mitochondrial dysfunction and cardiomyopathy, indicating a fundamental role of mitochondrial dynamics in cardiac physiology.93 Alterations in mitochondrial size and morphology have also been observed in diabetic animals and patients.9495 Incubation with high glucose induces mitochondrial fragmentation in cardiomyocytes by modulating phosphorylation and O-GlcNAcylation of Drp1 and Opa1.9697 Similarly, mitochondrial fragmentation and O-GlcNAcylation of Drp1 have been observed in diabetic hearts.9698 Increased mitochondrial fission may however also be mediated by lipotoxic effects in DC. Lipid-induced mitochondrial ROS have been shown to induce mitochondrial fission via post-translational modification of AKAP121, DRP1, and OPA1 in a mouse model of lipotoxicity.99
Dysfunctional mitochondria may be removed within cardiomyocytes by selective autophagy termed mitophagy. If mitophagy functions normally, injured mitochondria that are dysfunctional and may harm the cell (e.g. by increasing ROS) are removed, thereby protecting the heart. In contrast, defective mitophagy may result in accumulation of dysfunctional mitochondria and cause cardiac injury. Mitophagy requires to recruit the same core machinery as in general autophagy, but additionally recruits PINK, Parkin and a number of adaptors and receptors which specifically mediate mitophagy. In diabetic hearts, reduced protein levels of PINK and Parkin have been reported, and in high fat fed mice, impaired mitophagy via depletion of Atg7 or deletion of Parkin caused increased lipid accumulation, aggravated diastolic dysfunction and impaired systolic function, thus linking impaired mitophagy to the development of DC.87100101 Interestingly, ablating Drp1 interrupts mitochondrial fission and also upregulates Parkin, thereby causing mitochondrial depletion by mitophagy which contributes to the development of cardiomyopathy, implying that an interdependence between mitophagy and mitochondrial fission exists in the heart, and that mitophagy and mitochondrial fragmentation may be required for successful mitochondrial quality control in the heart.102 Finally, mitochondrial content, structure and function are regulated by mitochondrial biogenesis signaling, with peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) being the major transcriptional regulator. Increases in mitochondrial area, DNA, and PGC-1α signaling may support an increase in mitochondrial biogenesis in DC, although it remains unclear whether these mitochondria are actually fully functional.833 The interactions between mitochondrial dynamics, biogenesis and mitophagy in the heart and in DC remain to be defined more clearly but may be essential for the ability of the heart to deal with mitochondrial defects in cardiac disease induced by diabetes.
3. Novel mechanisms of mitochondrial dysfunction in DC
Mitochondrial dysfunction is a well known trait of DC both in human and animal diabetes and includes impaired mitochondrial respiratory capacity, increased mitochondrial oxidative stress, increased sensitivity for calcium-induced opening of the mitochondrial permeability transition pore, abnormal mitochondrial ultrastructure, transcriptional and translational downregulation of OXPHOS subunits, and impaired activity of Ca2+-sensitive dehydrogenases and the F0F1-ATPase.103 A novel mechanism potentially contributing to mitochondrial dysfunction but also to other alterations in DC may be posttranslational modifications, in particular increased mitochondrial protein lysine acetylation.104 Activity of sirtuin 3 (SIRT3), a NAD+-dependent mitochondrial deacetylase and major regulator of intramitochondrial protein acetylation, may be decreased in the diabetic heart, resulting in ROS accumulation due to increased acetylation and thus inhibition of Manganese superoxide dismutase.105 Furthermore, SIRT3 deficiency seems to aggravate suppression of autophagy and mitophagy in the diabetic heart, whereas SIRT3 overexpression activated autophagy and mitophagy, attenuated mitochondrial defects and decreased cardiomyocyte apoptosis.106 Decreased SIRT3 activity in DC may result from NAD+ depletion due to increased PARP-1 activity, thus depleting the necessary cosubstrate for SIRT3, or as a consequence of preexistent SIRT3-independent mitochondrial dysfunction, which may also lead to an increased NADH/NAD+ ratio and subsequent inhibition of SIRT3, thereby further aggravating mitochondrial defects.107 Since lack of SIRT3 in the heart leads to mitochondrial dysfunction, slowing of mitochondrial oxidative pathways, cardiac hypertrophy and increased fibrosis, the contribution of decreased SIRT3 activity to DC may be quite of significance.108
Other emerging mechanisms contributing to mitochondrial dysfunction in DC include increased ROS generation by monoamine oxidases (MAO) and mitochondrial calpains (e.g. Capn1).109 MAO-A and MAO-B are mitochondrial flavoenzymes that generate H2O2 during deamination of catecholamines, serotonin and biogenic amines, and their expression is increased in diabetic hearts.110 Inhibition of MAO activity using clorgyline or selegiline decreased ROS production in diabetic hearts by 50% and may represent a novel and promising approach to attenuate oxidative stress in DC.110 Furthermore, initiation of mitochondria-associated endoplasmatic reticulum membranes (MAMs) through FUN14 domain containing 1 (Fundc1) has been shown to result in increased mitochondrial Ca2+ content and mitochondrial fragmentation.111112 Fundc1 is an outer membrane protein which shows elevated levels in heart tissue of diabetic patients and induces MAM formation.111 High levels of glucose inhibit AMPK activity while overexpression of AMPK attenuates the mitochondrial dysfunction by ablating MAM formation implicating that Fundc1 suppression through AMPK might be a way to attenuate mitochondrial dysfunction in DC.111 Finally, mitochondrial oxidative stress in DC may result from mitochondrial accumulation of the Ca2+-dependent thiol protease calpain 1, which may decrease ATP synthase content and activity by cleavage of ATP synthase subunits, thereby impairing ATP regeneration and increasing mitochondrial superoxide generation.109 Mechanisms of mitochondrial dysfunction, as well as the potential interrelation between mitochondrial dysfunction, alterations in mitochondrial dynamics and mitophagy are schematically illustrated in Fig. 3.
Fig. 3
Novel mitochondrial mechanisms potentially contributing to DC. Decreased SIRT3 activity in DC may contribute to impaired oxidative ATP regeneration, increased ROS production and suppression of mitophagy. MAO and calpain 1 may contribute to increased mitochondrial ROS. O-GlcNAcylation and acyl-CoA driven posttranslational modification of Drp1 and Opa1 may contribute to mitochondrial fragmentation. Decreased levels of PINK and Parkin may suppress mitophagy in DC. Mitochondrial fragmentation and suppression of mitophagy may further amplify mitochondrial dysfunction in DC.
DC, diabetic cardiomyopathy; SIRT3, sirtuin 3; ATP, adenosine triphosphate; ROS, reactive oxygen species; MAO, monoamine oxidases; O-GlcNAc, O-linked β-N-acetylglucosamine; TCA, trichloroacetic acid.
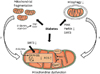
4. O-GlcNAcylation in DC
Another glucose-induced modification in diabetes besides AGE formation that currently attracts increasing attention is glycosylation of proteins via addition of an O-linked β-N-acetylglucosamine (O-GlcNAc), termed O-GlcNAcylation.113 Increased flux through the hexosamine pathway in diabetes increases the generation of UDP-GlcNAc, which is used by O-GlcNAc transferase to catalyze the addition of O-GlcNAc to proteins. Delivery of O-GlcNAcase to diabetic hearts reduced overall O-GlcNAcylation, restored Ca2+-handling and sensitivity of myofilaments to Ca2+, and improved cardiac contractility, indicating that excessive O-GlcNAcylation is detrimental for the heart.114 An effect on Ca2+ handling is also mediated by O-GlcNAcylation of CaMKII, which increases spontaneous SR Ca2+ release, thereby contributing to contractile dysfunction and cardiac arrhythmias.76 Increased O-GlcNAcylation of the mitochondrial fusion protein OPA1 or the mitochondrial fission protein Drp1 may lead to mitochondrial fragmentation, may impair mitochondrial membrane potential and impair complex IV activity.9697 Recently, it was reported that an interaction between O-GlcNAc transferase and complex IV, as occurs in healthy mitochondria, is impaired in mitochondria from diabetic hearts. Interestingly, the same authors also provided evidence for the existence of a UDP-GlcNAc transporter for mitochondrial UDP-GlcNAc uptake, which may suggest a significant regulatory role of this posttranslational modification in mitochondrial dysfunction in DC.115 O-GlcNAcylation may also increase fibrosis in DC by increasing levels of TGF-ß1 and downstream SMAD proteins.116
5. Micro-RNAs (miRNAs) in DC
miRNAs are noncoding single-strand RNAs of short length that bind to specific nucleotide sequences in the 3′ untranslated region of a target gene and suppress target protein expression by promoting degradation or repressing translation of target messenger RNA (mRNA). The significance of miRNAs for cardiac physiology has been emphasized by studies in mice with cardiomyocyte selective deletion of dicer, which impairs miRNA biogenesis and leads to dilated cardiomyopathy.117 In DC, a dysregulation of 316 out of 1,008 total miRNAs was observed, and pathway analysis implicated a number of miRNAs in apoptosis, oxidative stress, autophagy and cardiac hypertrophy.118 A number of studies investigated the actual contribution of specific miRNAs to DC. Proviral integration site for Moloney murine leukemia virus-1 (Pim-1) is a serine/threonine kinase which regulates mitochondrial integrity, apoptosis and cellular Ca2+ handling by changing the expression and/or activity of Bcl-2 family proteins and SERCA2a. Pim-1 is a direct target of miR-1, and increased miR-1 levels may impair Pim-1 expression in DC. Adenovirus-mediated rescue of myocardial Pim-1 expression in vivo improved diastolic and systolic function, attenuated ventricular dilation, attenuated fibrosis and apoptosis, and restored SERCA2a content in DC.119 Expression of miR-133 is increased in DC, and miR-133 exerts direct inhibitory effects on collagen production by impairing connective tissue growth factor expression, suggesting that increased miR-133 levels may promote fibrosis in DC.120121 Therapeutic silencing of increased miR-195 levels has been shown to reduce myocardial hypertrophy and to improve coronary blood flow and myocardial function in diabetes, maybe by reducing oxidative stress, inhibiting apoptosis and promoting angiogenesis, possibly by increasing reduced expression of Bcl-2 and SIRT1.122 Myocardial expression of miR-451 is markedly increased in mice fed a high-fat diet, and cardiomyocyte-specific deletion of miR-451 attenuates cardiac fibrosis, cardiac hypertrophy, and ceramide accumulation in this model. Attenuation of hypertrophy may result from restoration of decreased AMPK activity, which in turn may normalize increased mTOR phosphorylation and thus prevent high-fat diet induced cardiomyocyte growth.123 Expression of miR-30c is decreased in DC, and restoration of miR-30c levels attenuates cardiomyocyte hypertrophy, likely by normalizing increased levels of the pro-hypertrophic proteins cell division control protein 42 homolog (Cdc42) and P21 protein (Cdc42/Rac)-activated kinase 1 (Pak1).124 Finally, upregulation of miR-30d in DC was proposed to decrease FoxO3a signaling, resulting in caspase 1 activation, increased inflammatory signaling and thus pyroptosis.125 Based on the various traits and mechanisms of DC that may be regulated by miRNAs, a significant contribution to the development of DC has been proposed.126
6. Epigenetic mechanisms in DC
The term epigenetics describes heritable mechanisms that regulate gene expression independent of changes in DNA sequence. Epigenetic mechanisms generally include histone modification, DNA methylation and accelerated degradation of telomeres. Histone acetylation is achieved by the activity of histone acetyltransferases (HATs), whereas histone deacetylation is mediated by histone deacetylases (HDACs). Inhibition of HDACs has been shown to blunt pressure overload-induced cardiac hypertrophy and to attenuate myocardial ischemia reperfusion injury, thus indicating the significance of this epigenetic mechanism in cardiac disease.127128 Recently, it was reported that inhibition of HDAC activity with sodium butyrate resulted in attenuation of cardiac dysfunction, cardiac hypertrophy, fibrosis and apoptosis in DC of type 1 diabetic mice, indicating that epigenetic regulation also significantly contributes to DC.129 Furthermore, incubation of H9c2 cardiomyocytes in high glucose medium leads to decreased expression of insulin-like growth factor 1 receptor, which is prevented by inhibition of HDAC activity.130 HDAC inhibition also normalizes decreased AMPK phosphorylation, and increased expression of PGC-1α, cluster of differentiation 36, diacylglycerol O-acyltransferase 1 and 2, TNFα, and IL-6, accompanied by reversal of mild cardiac dysfunction and dilation.131 Finally, histone 3 lysine-9 tri-methylation seems to be responsible for a prolonged increase in cardiomyocyte IL-6 expression even after removal of high glucose from the medium, whereas apoptosis and mitochondrial dysfunction rapidly normalized.132 This observation suggests that epigenetic regulation may even contribute to detrimental effects of hyperglycemia that continue after normalization of glucose levels, a challenge in diabetes treatment termed metabolic memory.
7. Alternative splicing in DC
Alternative splicing is a physiological process that increases the diversity of proteins by variable inclusion or exclusion of exons during mRNA transcription. Known regulators of alternative splicing include the RNA binding proteins CUGBP Elav-like family member 1 (CELF1) and RNA binding fox-1 homolog 2 (RBFox2). Overexpression of CELF1 or deletion of RBFox2 causes splicing defects and cardiomyopathy, emphasizing the significance of alternative splicing for myocardial integrity and function.133134 In DC, alternative splicing reverts to an embryonic pattern, mediated by activation of protein kinase C α/β and subsequent phosphorylation of CELF1 and RBFox2.135 In addition, expression of a dominant-negative isoform of RBFox2 protein in early DC, which suppresses activity of the native endogenous RBFox2 protein, alters alternative splicing in the heart, resulting in impairment in Ca2+ handling and excitation-contraction coupling.134136
THERAPEUTIC POSSIBILITIES IN DC
The established as well as the emerging molecular mechanisms of diabetic cardiomyopathy are potential candidates for a therapeutic intervention. Antioxidant treatment to prevent ROS-associated damage is one of the therapeutic strategies that could be beneficial in DC. Several mitochondria-targeting antioxidative compounds such as MitoQ, MitoVitE, or MitoTempol are available. In particular, treatment with MitoQ showed beneficial effects in different pathological conditions such as hypoxia, acute endotoxemia, ischemia-reperfusion or pressure overload in rodent models.137138139140 Although none of these compounds have been tested for treatment of DC yet, treatment with MitoQ decreased mitochondrial ROS production, polymorphonuclear neutrophils (PMN) rolling, PMN adhesion, NF-κB and TNFα while increasing levels of glutathione peroxidase 1 and PMN rolling velocity in leukocytes of type 2 diabetes patients, indicating anti-inflammatory and antioxidative effects and suggesting a potential benefit in DC.141
Another potential therapeutic strategy may be activation of sirtuins to maintain mitochondrial function in DC by increasing the NAD+/NADH ratio. Nicotinamide mononucleotide (NMN) and nicotinamide riboside (NR) are NAD+ precursors which increase the cellular levels of NAD+ in animal models, including the heart.142143144 Recently, it has been shown that oral supplementation with NR could increase NAD+ levels and is well tolerated in healthy humans.145 In a model of heart failure due to pressure overload, treatment with NMN maintained cardiac function.146 Another pharmacological strategy to maintain the NAD+/NADH ratio may be to inhibit PARP-1 using INO1001, which has been shown to ameliorate oxidative stress, inflammation and fibrosis in hearts of type 2 diabetic mice.147 PARP-1 inhibitors are primarily used as drugs against multiple cancers and were first approved 2014 by the Food and Drug Administration.148
Restoring metabolic flexibility in the diabetic heart represents another therapeutic intervention to improve cardiac function in DC. There are various agents to alter cellular uptake and/or oxidation of energy substrates with the goal to restore the balance between fatty acid utilization and glucose metabolism. Trimetazidine and ranolazine both inhibit fatty acid oxidation. Trimetazidine improved cardiac function in diabetic patients with idiopathic cardiomyopathy, while ranolazine improved interventional hemodynamic measurements but not relaxation parameters in HFpEF patients.149150 Perhexiline, Amiodarone and Etomoxir inhibit carnitine palmitoyltransferase 1 and thereby reduce fatty acid oxidation.151 Perhexiline improved maximal oxygen uptake, left ventricular ejection fraction, myocardial function, and skeletal muscle energetics in chronic heart failure patients and also cardiac energetics in patients with dilated cardiomyopathy.152153 More studies in animal models and humans are required to further evaluate the effects of restoring metabolic flexibility as a therapeutic option in DC.
Antidiabetic drugs, by their various mechanisms of action, may certainly also treat different underlying mechanisms of DC, in particular including sodium-glucose cotransporter-2 inhibitors. This topic has been comprehensively addressed in recent reviews and will thus not be discussed in this manuscript.154155
CONCLUSIONS
Diabetes is a steadily growing epidemic, and cardiovascular death is the main cause of morbidity and mortality in these subjects. DC is increasingly recognized by physicians as a cause of cardiac deterioration despite non-significant CAD and hypertension, and as a significant contributor to the development of both HFpEF and HFrEF. As reviewed above, several mechanisms have been established in the past that may significantly contribute to DC, with increasing new insights into underlying mechanisms in recent years. In addition, novel and emerging mechanisms have been identified, including removal of damaged cellular components (autophagy, mitophagy), or transcriptional (FoxO1, alternative splicing, miRNAs, epigenetics) and posttranslational regulation (O-GlcNAcylation, protein deacetylation) of proteins. Further understanding of DC and the aforementioned mechanisms is certainly imperative for potential development of promising drugs for DC in the future. Having said this, it is equally important to already initiate studies translating promising targets and successful therapeutic interventions observed in small animal models into larger animal models or even into humans.
 XML Download
XML Download