

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
The gastrointestinal tract harbors 10–100 trillion microorganisms composed of bacteria, viruses, fungi, and parasites. The microbial unique gene set contains more than 150 times the number of genes than are present in the host genome [1]. Bacteria comprise more than 99% of microorganisms in the gut and between 1,000 and 1,150 species have been reported to be present in the gut with at least 160 different species reported in individuals [1]. Gut microorganisms play important roles in the body, with mutual interactions between the host and microbes influencing the innate and acquired immunity, nutrient absorption, vitamin synthesis, drug metabolism, cardiac size, and host behavior [23]. Many factors, such as age, antibiotic use, psychological stress, radiation, race, gender, hygiene, host genetics, and diet, cause changes in the gut microbiome community, inducing significant interpersonal and intrapersonal diversity in the gut microbiome [4]. Detrimental changes in the microorganisms, called dysbiosis, have harmful effects on the host species, which have been linked to allergies, asthma, obesity, neurological disorders, cardiovascular disorders, diabetes, and enteritis [567].
Fecal microbiomes play a critical role in obesity in humans and animals. Adult germ-free mice exposed to the cecal contents collected from conventional normal mice increased the level of fat storage by 60% and increased insulin resistance within 14 days despite the decreased dietary intake [8]. The gut microbiota also influence the energy harvesting capacity from the enteric contents in metabolic and biochemical analyses, which is transmissible by microbiota transplantation [9]. The mechanisms through which the microbiome induces obesity include altered short chain fatty acid production, altered host gene expression, and host inflammation [1011]. The relative proportion of phylum-level bacterial representation has focused on obesity in human and animals [1213]. A higher abundance of Firmicutes and lower abundance of Bacteroidetes have been observed in obese humans and mice compared to their lean counterparts [91214]. In addition, a higher representation of Actinobacteria, lower Bacteroidetes, and no significant difference in Firmicutes have been found in human populations [13]. A decrease in bacterial diversity has been associated with obesity in previous reports, with diet being the main determinant of the gut microbiome composition and diversity [12151617]. A western style diet is characterized by higher fat and protein contents and a lower fiber content, which selects for differences in bacterial representation as assessed at the phylum level [18]. The phylum Bacteroidetes was shown to be the dominant genus represented in long-term high protein and animal fat diet groups, whereas the genus Prevotella was enriched in the high carbohydrate diet groups [1719].
Non-human primates (NHPs) are the most valuable animal models for biomedical studies. The gut microbiomes of NHPs have been evaluated widely [18202122] and found to be distinct from those of humans based on differences in the host species and diet adaptation [182023]. The macaque microbiome harbors Bacteroidetes, Firmicutes, and Proteobacteria similar to the human microbiome, but Spirochetes and Helicobacter were significantly abundant compared to humans [23]. A recent report characterized the differences between captivity-humanized NHP microbiomes and wild state NHPs [20]. Prevotella and Bacteroides were shown to be the dominant genera in the NHP gut microbiomes under captive circumstances, which is similar in composition to the human microbiomes [20].
The results reported in the literature regarding the associations between gut microbiome composition and obesity are inconsistent because of the inaccuracy of human retrospective and intervention studies and differences in animal species [10]. To the best of the authors' knowledge, no studies have evaluated the association between the gut microbiome and obesity in the NHP model. This study compared the microbiome communities of obese and lean groups of captive cynomolgus monkeys using the 16s rRNA sequencing method. All animals were imported from China and reared under identical environmental conditions, including temperature, humidity, light and diet. The medical history of the animals was identified; therefore, the relatedness of obesity and microbiomes could be understood precisely under the strict controlled conditions of this study.
MATERIALS AND METHODS
Animals and environments
A total of seventeen female cynomolgus monkeys (Macaca fascicularis), ranging in age from eight to ten years, were selected and divided into two groups based on their body weight, obesity (n = 8, average body weight = 4.26 ± 0.39 kg), and lean (n = 9, average body weight = 2.62 ± 0.11 kg) groups (Table 1). The body weight proved to be the key determinant for obesity in captive female cynomolgus monkeys in a previous report [24]. One monkey (C032) in the lean group showed chronic diarrhea without the presence of pathogenic bacteria and parasites causing chronic diarrhea in monkeys, including Campylobacter jejuni, Clostridium difficile, Salmonella spp., Shigella spp. and Yersinia enterocolitica (unpublished data). The difference in body weight between the obese and lean groups was statistically significant (p < 0.001). All animals, which originated from China, were reared in individual indoor cages at the National Primate Research Center (Korea Research Institute of Bioscience and Biotechnology, Korea). They were fed commercial monkey feed (2050 Teklad Global 20% Protein Primate Diet; Harlan, Envigo, UK) consisting of 20% crude protein, 5.4% fat, 8.1% crude fiber, and 40.1% carbohydrate, as well as various fruits according to season, and water ad libitum. The environmental conditions were set automatically at a temperature of 24°C ± 2°C, relative humidity of 50% ± 5%, light intensity of 150–300 Lux, ventilation at 10–20 times per an hour, and a 12 light/12 dark hour cycle. All animal housing conditions satisfied the minimal requirements as outlined in ‘The Guide for the Care and Use of Laboratory Animals’ published by the Institute for Laboratory Animal Research in 2010. Veterinary diagnoses and care were performed by institutional veterinary experts. The monkeys had no medical history, including antibiotic use, within at least 4 weeks. All procedures were approved by the Institutional Animal Care and Use Committee of Korea Research Institute of Bioscience and Biotechnology (IACUC number: KRIBB-AEC-18144).
Table 1
Information of cynomolgus monkeys evaluated in this report

Total DNA preparation
Fecal samples were collected from all monkeys at the same time. The samples were placed in sterile conical tubes, frozen immediately, and stored at −80°C until processing. The frozen samples were thawed on ice and the internal portion of the fecal matter was then collected aseptically. The samples were weighed and placed into 2 mL microfuge tubes. The total DNA was extracted from each fecal sample using the QIAamp® DNA stool Mini Kit (Qiagen, USA) according to the manufacturer's instructions.
Polymerase chain reaction (PCR) amplification of the bacterial 16S rRNA gene
The V3 to V4 hyper-variable region within the 16S bacterial rRNA gene was amplified from the extracted fecal DNA with two primers: 341F sense primer (5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG-3′) and 805R antisense primer (5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHV GGGTATCTAATCC-3′). The thermal cycling conditions were as follows: initial denaturation at 95°C for 30 sec followed by 25 cycles of denaturation at 95°C for 30 sec, primer annealing at 55°C for 30 sec, and extension at 72°C for 30 sec followed by a final extension step at 72°C for 5 min. Secondary PCR was performed to attach the Illumina NexTera barcode using the i5 sense primer 5′-AATGATACGGCGACCACCGAGATCTACACXXXXXXXXTCGTCGGC AGCGTC-3′ and i7 antisense primer 5′-CAAGCAGAAGACGGCATACGAGATXXX XXXXXGTCTCGTGGGCTCGG-3′, with the barcode regions indicated by the X positions. The secondary PCR conditions were the same as described above except that eight amplification cycles were used. The PCR products were separated by electrophoresis on 1% agarose gels and visualized using a Gel Doc system (BioRad, USA). The targeted amplicons were pooled and purified using the CleanPCR system (CleanNA, The Netherlands). The amplicon quality and size were evaluated using a DNA 7500 chip on a Bioanalyzer 2100 system (Agilent, USA). Sequencing of target-sized amplicons was performed at ChunLab, Inc. (Korea) with an Illumina MiSeq Sequencing system (Illumina, USA) according to the manufacturer's instructions.
MiSeq pipeline
To improve the data quality, low quality (< Q25) reads were filtered using the Trimmomatic 0.32 read trimming tool [25]. Among the quality-controlled raw data, paired-end sequence data (250 bp) were assembled using PANDAseq [26]. The primers were trimmed at a similarity cut-off of 0.8 using a ChunLab in-house program. Non-specific amplicons encoding non-16S rRNA genes were detected using the HMMER hmmsearch program [27] and were not evaluated for further sequence analysis. The sequences were denoised using DUDE-Seq to correct for sequencing errors [28] and the non-redundant reads were finally extracted using UCLUST-clustering [29]. Bacteria taxonomic assignments were performed based on the EzBioCloud database using USEARCH (8.1.1861_i86linux32) [29] and sequence similarity calculations via a precise pairwise alignment [30]. The species level taxonomic assignments were confirmed based on the presence of reference sequences over the 97% similarity cut-off values in the EzBioCloud database. For sequences with less than 97% similarity, the chimeric sequences were identified and eliminated for further analysis based on UCHME [31] and the non-chimeric 16S rRNA database from EzBioCloud. The non-chimeric sequences were clustered based on the cluster database at high identity using the tolerance [32] and UCLUST tools with 97% similarity cut-off values. The range of sequence similarities in taxonomy were as follows: genus (97 ≥ X ≥ 94.5), family (94.5 ≥ X ≥ 86.5), order (86.5 ≥ X ≥ 82), class (82 ≥ X ≥ 78.5), and phylum (78.5 ≥ X ≥ 75).
Data analysis
Pyrosequencing data was analyzed using the CLcommunity™ program, version 3.46. (ChunLab, Inc.). Bacterial taxonomic composition was evaluated from the phylum to species level. The sharedness between the two groups was examined using a source tracking method. To evaluate the richness and diversity of gut microbiomes, the values of alpha diversity were examined, including the rarefaction curves and diversity indices. The indices included the values of valid reads, operational taxonomic units (OTUs), abundance-based coverage estimator (ACE), Chao1, Jackknife, Shannon, and Simpson. To compare the gut microbiome compositions of the obese and lean groups, beta analysis, including principal coordinate (PCO) analysis, was carried out based on fast UniFrac analysis [33]. The core microbiota was defined as individual bacterial phyla comprising over 0.1% in gut microbiomes. The enterotypes were classified based on the core microbiota. Statistical analysis was performed using unpaired t-tests using GraphPad PRISM 5.03 software (GraphPad Software, Inc., USA). The data are expressed as the mean ± standard deviation (SD). Statistical significance was determined as p values under 0.05.
RESULTS
Pyrosequencing analysis of fecal microbiomes
A total of 17 fecal samples derived from monkeys were obtained successfully and analyzed using Illumina sequencing techniques (Table 1). In total, 1,151,741 valid reads and 40,188 OTUs were obtained from 17 samples and used for further analysis. The average valid reads were 66,537.88 ± 20,452.24 (mean ± SD) in the obese group and 68,826.44 ± 44,355.34 (mean ± SD) in the lean group. The average OTUs were 2,167.37 ± 762.49 in the obese group and 2,538.78 ± 1,330.20 in the lean group. There were no significantly different values of valid reads or OTUs between the two groups.
Microbial compositions
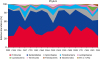
Comparative taxonomic composition of the two groups were analyzed from the species to phylum level. At the phylum level, a higher composition of Spirochetes (p = 0.036) was observed in the lean group (Table 2). No significant differences in Firmicutes or Bacteroidetes phyla were observed between the two groups (Fig. 1). Different values of Spirochetes (p = 0.036) at the class level, Spirochaetales (p = 0.036) and Burkholderiales (p = 0.042) at the order level, Spirochaetaceae (p = 0.035) and Sutterellaceae (p = 0.017) at the family level, Alloprevotella (p = 0.0456), Oligospaeara_uc_g (p = 0.050), DQ824928 (p = 0.018), and Sutterellaceae_uc (p = 0.046) at the genus level were identified. The rank abundance of bacteria composition in the monkey gut microbiomes were analyzed and are listed in Table 2. In source tracking analysis, the average values of sharedness between the two groups were 98.62%, 99.17%, 99.89%, 99.95%, 99.99%, and 99.99% at the species, genus, family, order, class, and phylum levels, respectively. Higher sharedness values (95.81% ± 2.28% at the genus level in chart A, 79.54% ± 5.88% at the species level in chart B) were observed among individual monkeys regardless of their body weight, but these relative rates of bacteria composition are represented as a variable (Fig. 2). No exclusive bacteria at the phylum to species levels were found between the groups based on taxon exclusive XOR analysis. Significant variable bacterial compositions were found at the phylum level among the individual monkeys (Fig. 3).
Table 2
Core gut microbiomes of cynomolgus monkeys of obese and lean groups at a phylum level

Fig. 1
Relative abundance of the major bacterial phyla in obese and lean groups of cynomolgus monkeys. Seven major bacterial phyla, (A) Firmicutes, (B) Bacteroidetes, (C) Spirochaetes, (D) Proteobacteria, (E) Lentisphaerae, (F) Cyanobacteria, and (G) Tenericutes were identified. The differences in relative abundance between obese and lean groups were analyzed statistically using one-way analysis of variance.
*Statistically different values (p < 0.05) between two groups were observed in Spirochaetes at the phylum level.


Diversity analysis of microbiome samples
The number of OTUs was similar in the two groups (obese group: 2,167.375 ± 762.4888, lean group: 2,538.778 ± 1,330.204, p = 0.499). The bacterial richness estimates and diversity indices in samples were calculated. No significant differences in estimated species richness estimates (Chao1 and ACE) and diversity indices (Shannon and Simpson) were identified between the two groups (Fig. 4). The average value of the species richness estimates was 2,189.77 ± 763.18 for the obese group and 2,565.54 ± 1,329.53 for the lean group in ACE analysis; 2,170.05 ± 762.05 in the obese group and 2,126.52 ± 1,329.30 in the lean group in Chao1 analysis; and 2,230.5 ± 766.73 in the obese group and 2,612.89 ± 1,333.73 in the lean group in the Jackknife indices. Species diversity by Shannon indices analysis of the obese and lean groups were 4.98 ± 0.41 and 5.31 ± 0.22, respectively. The Simpson index of the obese and lean groups were 0.03 ± 0.01 and 0.02 ± 0.01, respectively.

Comparison of microbiome diversity between the obese and lean groups
Fast UniFrac analysis was performed to compare the microbiome composition between the two groups. Distinct clustering between the groups was not observed in PCO analysis using the unweighted pair group method with an arithmetic mean based on the UniFrac distance (Fig. 5). A distinct Bacteroidetes-rich group was identified.

DISCUSSION
Recent studies focused on the role of the gut microbiomes in obesity. On the other hand, there are inconsistent results regarding the relationship between obesity and microbiome composition in animals and humans because of the inaccuracy of human retrospective and intervention studies, and animal species differences [1013]. Recently, captive NHPs proved to harbor humanized gut microbiomes [34]. In this report, the fecal microbiomes were analyzed comparatively between obese and lean groups of captive cynomolgus monkeys that had been reared in individual cages under identical environmental conditions.
The Bacteroidetes, Firmicutes, and Proteobacteria phyla were dominant in the fecal microbiomes similar to human microbiomes (Table 2). In addition, Actinobacteria and Verrucomicrobia were found at the phylum level in the obese groups, which are rare in NHPs [2335]. All animals commonly harbored abundant amounts of the genus Prevotella, but not Bacteroides. These findings suggest that captive cynomolgus monkeys have more similar characteristics of the gut microbiomes to humans compared to other NHP studies [2335]. In general, the characteristics of obesity-related microbiomes revealed an increased relative proportion of Firmicutes and Bacteroidetes and decreased bacterial diversity [10]. On the other hand, some studies failed to verify the phylum level changes and decreased bacterial diversity [101236]. In this report, no significant differences in the relative proportion of Bacteroidetes and Firmicutes were found. At the phylum level, only the increased levels of Spirochetes were statistically significant (p = 0.036) in the lean group compared to the obese group (Fig. 1). Spirochetes are part of the normal gut microbial components in NHPs, which is absent from typical human microbiomes [23]. This result strongly suggests that the relative proportion of Firmicutes and Bacteroidetes is questionable as a factor inducing obesity in mammalian species and could be the result of a range of factors related to obesity, particularly the diet, because high fat diets have been shown to increase the relative abundance of Firmicutes without obesity in animals [3738].
Lower bacterial diversity has been generally identified in obese people compared to their leaner counterparts [10]. Previous reports found that dietary modification changed gut bacterial diversity in the host significantly [1518] and that the determinants that affected the bacterial diversity included phylogeny, age, captivity, and nutrition [102139]. In this report, the species richness and diversity values varied according to the individual, regardless of obesity, even though the all monkeys in this study were of similar age and were reared under the same environmental and diet conditions over a long period of time (Fig. 4). Other factors in addition to those mentioned previously might have affected the gut bacteria diversity considering the inter-individual variations of bacterial diversity. Distinct clustering according to obesity was not identified by PCO analysis (Fig. 5), even though long-term dietary patterns have been shown to affect the gut microbial enterotypes directly [17]. Interestingly, distinct clustering of three samples was observed that was different from the other samples, which all belonged to the obese group, even though all animals were fed an identical diet. This group harbored more Bacteroidetes.
Gut microbiome communities are influenced by many factors, including age, antibiotic use, psychological stress, radiation, race, gender, host genetics, and diet, showing significantly high interpersonal and intrapersonal diversity in the gut microbiomes [4]. Therefore, only approximately 38% of the total gut microbial genes are shared among humans [1] and at the species level, 70% of phylotypes are unique to each person [12]. Under identical environmental conditions, significantly higher levels of bacterial species sharedness were observed compared to previous reports (Fig. 2). On the other hand, the relative abundance of bacterial components was highly variable in individuals irrespective of obesity (Fig. 3). Therefore, environment factors, particularly diet, appear to have a profound effect on the gut microbial components rather than abundances.
To the best of the authors' knowledge, this is the first study to characterize the fecal microbiomes related to obesity in captive cynomolgus monkeys and fecal microbiome composition under strict identical environmental conditions. No significant differences in relative proportion of Bacteroidetes and Firmicutes were found between two groups. These proportions of bacterial phyla itself do not appear to be a critical factor inducing obesity in the body. All animals showed significantly higher species sharedness, but varying relative abundances of these bacterial components. Therefore, environmental factors, including diet, strongly affect the bacterial species components rather than abundance. In addition, NHPs were found to be a valuable preclinical animal model for human microbiome studies based on the microbiome composition.
 XML Download
XML Download