

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Psoriasis is a chronic, recurrent, inflammatory skin disease characterized by prominent epidermal hyperplasia and a distinct inflammatory infiltrate. The estimated prevalence of this cutaneous disease is between 0.5% and 3.15% of the U.S. population.1234 The disease pathogenesis results from the interplay between immunocytes and keratinocytes and the resulting production of cytokines, chemokines, and growth factors. Several different forms of psoriasis have been described such as inverse psoriasis, pustular psoriasis, guttate psoriasis, and psoriasis vulgaris. Psoriatic arthritis is a distinct inflammatory arthritis seen in conjunction with psoriasis, but with varied timing and genetic control. Most psoriasis patients develop a chronic ‘plaque-type’ psoriasis referred to as psoriasis vulgaris through a primarily T helper (Th)17/cytotoxic T (Tc) 17-type immune profile.56 Although these cytokine profiles control the infiltrative T cells of psoriatic lesions and the epidermal response, the main factors responsible for plaque formation and disease severity are yet to be fully distinguished. As antigen-specific T-cell activation is considered crucial in the development of the disease, it is hypothesized that antigen presentation in the skin might be linked to the activation of specific T-cell subsets at the site. In psoriasis vulgaris, novel autoantigens, LL-37 and a disintegrin like and metalloprotease domain containing thrombospondin type 1 motif-like 5 (ADAMTSL5), have recently been identified as stimuli in the Th17/Tc17 activation of psoriatic lesions.567
In early lesions of human psoriatic skin, the characteristic dermal changes include tortuous dilated blood vessels, papillary edema, and perivascular infiltration composed of dendritic cells (DCs), lymphocytes, and macrophages. In chronic lesions, such as those in patients with plaque psoriasis, the epidermal changes become more evident which include thickening (acanthosis), elongation of rete ridges, and finally elongation of the dermal papillae which contain dilated capillaries. Proliferation of keratinocytes predominates in both the basal and suprabasal layers of the epidermis. As such, disturbing the differentiation process of keratinocytes can ultimately reduce thickness of the stratum granulosum of the epidermis. This results in irregularly thickened stratum corneum (hyperkeratosis) and retention of the nuclei inside corneocytes (parakeratosis).8 Unlike other inflammatory skin diseases, infiltrating inflammatory cells in the dermis of psoriatic lesions include macrophages, neutrophils, and lymphocytes. In these lesions, CD4+ T cells represent the majority of lymphocytes localized to the dermis, while CD8+ T cells reside primarily within the epidermis.8910 In pustular variants especially, neutrophil infiltration within the stratum corneum and adjacent epidermal layers is a diagnostic histopathological feature.8
Psoriasis is a uniquely human condition, but numerous genetic approaches to model the disease in mice have been attempted. Most of the mouse models of psoriasis-like disease show the histopathological features that closely resemble those observed in human psoriasis, though the pattern and type of immune cell infiltrate has yet to be exactly reproduced. Current mouse models only capture psoriasis-like skin disease with plaque-like lesions that have features such as hyper keratosis and/or parakeratosis, infiltration of T cells within the epidermis, neutrophilic microabscesses, enhanced vascularization, and finally involvement of joints and nails similar to psoriatic arthritis.8910
Despite therapeutic advances, complete remission in psoriasis patients is not achievable, and relapse often occurs within weeks or months. In this review, we describe the current knowledge about the emerging role of synthetic antimicrobial peptides (AMPs), TRAIL and NRP1 blocking peptides in the pathogenesis of psoriasis and the potential for identifying new drug targets with better efficacy and less adverse effects.
CURRENT TREATMENTS
A multitude of topical and systemic treatments, including corticosteroids, vitamin D3 analogs, retinoids, methotrexate and cyclosporin, in addition to phototherapy with ultraviolet irradiation have been successfully used for the treatment of psoriasis. In the past decade, the development of biologic agents has shown promising therapeutic benefit for refractory cases of psoriasis. These biologic agent target either general inflammation through the TNF pathway, or specific immune cell activation through manipulation of CD2, IL-12, IL-17 and IL-23 expression.1112 These systemic agents that have been developed and approved to treat moderate-to-severe psoriasis include T-cell inhibitors (efalizumab and alefacept),1314 tumor necrosis factor (TNF-α) inhibitors (infliximab, adalimumab, and etanercept),151617 interleukin (IL)-12/17/23 inhibitors (secukinumab, ixekizumab, ustekinumab, and brodalumab),1819202122232425 and small molecule inhibitors such as the janus kinase inhibitor, tofacitinib, and the phosphodiesterase 4 inhibitor, apremilast.2627
Unfortunately, these drugs have potentially serious side effects and can predispose patients to the development of both infectious disease and malignancy due to immune modulation and suppression. Even with these strong therapeutic agents, relapse often occurs in patients. Therefore, it is increasingly important to identify new therapies with better efficacy and less adverse effects. Owing to the heterogeneity of psoriasis, future treatments are unlikely to be restricted to individual biological targets. Instead, it will be advantageous to focus on concurrent use of therapies to fight immune over activation and to repair differentiation mechanisms of epidermal keratinocytes at various stages of disease. It will also be advantageous to identify new drugs that can be used to wean patients off carcinogenic/immunosuppressive effects that were first used to obtain clinical responses.
PATHOGENESIS OF PSORIASIS
Although the cause of psoriasis is not fully understood, there are two main hypotheses regarding the etiology of disease development. The first considers psoriasis as primarily a disease of epidermal keratinocytes , in which disordered inflammation is also necessary for disease development. The second hypothesis is that the disease is an immune-mediated disorder in which the production of cytokines such as tumor necrosis factor-alpha TNFα, chemokines and growth factors by immunocytes and keratinocytes results in inflammation and epidermal hyper-proliferation. Once activated, T cells migrate toward the dermis and stimulate cytokine release which induces rapid production of skin cells. IL-17A and other cytokines act on keratinocytes to propel further inflammatory mediator production, thereby creating an inflammatory loop.
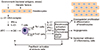
Skin homeostasis is regulated by communicating skin cell types as well as factors such as cytokines, chemokines and growth factors.28 This homeostasis can be interrupted by genetic alterations in the key signaling pathways of the immune system, provoking inflammation and psoriasis. Current mouse models have focused on keratinocytes, immunocytes, and especially T cells as disease initiators in the disease process.810 Psoriasis, previously considered a uniquely T-cell-dependent disease, is now thought to be more complex with keratinocytes playing a fundamental role in eliciting the initial pathogenic events and then sustaining the chronic phase of the disease.2930 Early in psoriasis, keratinocytes induce innate immunity and represent the “first-line responding” skin cells in the microenvironment.31 As shown in Fig. 1, keratinocytes mediate innate immunity though triggering pathways, such as skin trauma, pathogens (i.e., S. aureus), or drugs (Fig. 1). In psoriasis, stressed keratinocytes produce LL-37, which stimulates plasmacytoid DCs (pDCs) to produce IL-23, TNF, and interferon (IFN)-α. Keratinocytes also produce IL-1β, IFN-γ and TNF when under stress or dying, which activates DC to produce the proinflammatory cytokines and vascular endothelial growth factor (VEGF), ultimately resulting in vascular permeability and leukocyte adhesion to microvascular endothelium.32 Activated DCs migrate into the skin-draining lymph node and promote differentiation of the naive CD4+ and CD8+ T cells into IL-12-induced Th1 cells and IL-23-induced Th17 cells, respectively.33 Inflammatory cells, which are attracted by these keratinocytederived factors, migrate into the skin via lymphatic and blood vessels. Th1 cells interact with DCs on the dermis, which secrete cytokines such as IL-23 and TNF. These products then stimulate keratinocytes to activate NKT cells via the production of TNF and IFN-γ.34 In the dermis, Th17 cells secrete IL-17A/F, IL-21, IL-22, TNF and IFN-γ. The release of these cytokines prompts the production of IL-1β, IL-6 and other keratinocyte-derived cytokines and chemokines, which subsequently stimulate keratinocyte proliferation and impair keratinocyte differentiation.3435 In addition, Th1- and Th17-derived cytokines induce the release of β-defensins, the neutrophil chemoattractant and antimicrobial S100 proteins, and the neutrophil-recruiting CXC chemokine ligands (CXCL)-1, CXCL-3, CXCL-5 and IL-8 by keratinocytes. Together, these mediators perpetuate neutrophil infiltration into the epidermis, which is an important component of the pathogenesis of psoriasis.14 Keratinocytes also produce growth factors such as VEGF, FGF and angiopoietin 2 that promote an angiogenic response. The interaction between keratinocytes and inflammatory cells is mediated by cell products such as IL-1α, IL-6, IL-20, IL-21, IL-8 and transforming growth factor (TGF)-β. Fibroblasts release keratinocyte growth factor (KGF), epidermal growth factor (EGF) and TGF-β upon activation by these ligands and further promote epidermal proliferation, impaired differentiation, and the deposition and rearrangement of the extracellular matrix. Memory CD8+ T cells will bind to collagen IV in order to infiltrate the epidermis and contribute to the inflammation process.36 Furthermore, pathogenic inflammation results when chemokines attract NKT cells or T cells, which are then subsequently activated by glycolipid-CD1d complexes.37 As discussed above, keratinocyte-derived cytokines influence the growth of supporting stromal cells and vessels, which facilitate the access of inflammatory cells to the microenvironment.38 Crosstalk between the neighboring keratinocytes, leukocytes, and fibroblasts magnifies and sustains an inflammatory process, which is a hallmark of psoriasis.
IMPORTANCE OF KERATINOCYTES AND ITS CROSSTALK WITH IMMUNOCYTES IN PSORIASIS
The epidermis is largely composed of keratinocytes, which help initiate and maintain the skin's immune response and physical barrier.39 It also contains specialized cells such as melanocytes, Langerhans cells, and non-activated, dormant T cells. In psoriasis, keratinocyte stem cell proliferation pathways are dysregulated and aberrant, showing hyperactivation and hyper-proliferation of rapidly matured cells with decreased lipids and keratohyalin granules.4041
Stimuli such as trauma or infection can initiate an inflammatory response in genetically susceptible keratinocytes. The stimulated keratinocytes are capable of initiating an immune response through the production of various proinflammatory cytokines, which feedback on the keratinocytes, inducing further production of cytokines. Several antigens produced by stimulated keratinocytes, such as LL37 cathelicidin/nucleic acid complexes, newly generated lipid antigens, have been identified, and are suspected of inducing the initial activation of T cells. In turn, these Th1, Th17, and Th22 immune cells contribute to the development of psoriasis by secreting various cytokines including interferon γ (IFN-γ), tumor necrosis factor α (TNF-α), and interleukin 17 (IL-17). Thus, keratinocyte activation directly or indirectly contributes to the major histological features of psoriasis as discussed above. Although the identities of the specific cells that trigger the pathogenesis of psoriasis are unknown, mouse models have elucidated the roles of keratinocytes and T cells as disease initiators.8 Furthermore, it has been found that skin homeostasis can be interrupted by inflammatory cells due to genetic aberrations or abnormalities that affect key signaling pathways involved in inflammatory immune responses in keratinocytes, resulting in psoriasis. Thus, despite the current focus on the role of T cells, the keratinocytes affected by psoriasis are also clearly abnormal in many respects and likely overstimulate immunocytes via positive feedback of inflammatory cytokines and chemokines.29 One other notable abnormality in psoriatic keratinocytes is the overproduction of AMPs.42
INTRODUCTIONS OF MATERIALS FOR THERAPEUTICS OF PSORIASIS: SYNTHETIC AMPs, TRAIL AND NRP1 BLOCKING PEPTIDES
1. Synthetic antimicrobial peptides (AMPs)
Although psoriasis is classified as an autoimmune disease, considerable evidence suggests an important role for bacteria such as Staphylococcus aureus (S. aureus) in the pathogenesis of psoriasis. Several mutations in more than one gene have been implicated, and evidence suggests that environmental factors such as a common viral or bacterial infection or simple injury contribute to their activation and expression. Skin infections with S. aureus are thought to serve as initiation and/or persistence factors for numerous inflammatory skin diseases, including psoriasis.43 Colonization and/or infection with Staphylococcus and Streptococcus have been reported to exacerbate psoriasis and many other inflammatory skin conditions.4445 It has been theorized that toll-like receptors (TLRs) mediate this response through recognition of exogenous, microbial products and subsequent elicitation of a robust immune response. For example, VEGF-transgenic mice develop psoriasiform plaques that resemble human psoriasis. Peptidoglycan (PGN) from S. aureus induces expression of VEGF and subsequently IL-13 in keratinocytes, and IL-13 further induces VEGF expression via positive feedback. PGN induced VEGF and IL-13 expression was blocked by anti-Toll-like receptor 2 (anti-TLR2) antibody. These results suggest that PGN from S. aureus can further stimulate LL37 and VEGF expression in keratinocytes through TLR2, and that this VEGF production can be amplified by ensuing IL-13 overproduction.46 This implies that PGN from S. aureus is involved in the development of psoriasis via induction of VEGF expression.
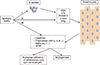
S. aureus has multiple mechanisms to modulate the immune system; one is through the production of proteins such as superantigen toxins, Staph protein A, and the cytolytic Staph α-toxin.43 Ezepchuk et al.47 found that S. aureus isolates from the skin of patients with atopic dermatitis and psoriasis were either characteristic superantigenic toxins or thermolabile toxins, secreting α-toxin and extracellular staphylococcal protein A. When superantigenstimulated immunocytes were injected into normal skin from subjects with psoriasis xenografted onto an immunodeficient mouse, clinical, histologic, and immunologic changes consistent with psoriasis were established.4849 Staphylococcal protein A, α-toxin, and superantigen toxins derived from S. aureus, which was isolated from patients with psoriasis and atopic dermatitis, can directly induce pro-inflammatory responses in keratinocytes through the release of TNF-α (Fig. 2). Superantigen (SAg), as an enterotoxin, activates keratinocytes through MHC-II molecules, resulting in the production of TNF-α as well as a calcium mobilization response. In contrast, S. aureus-derived protein A does not appear to activate a calcium mobilization response although it is a potent inducer of TNF-α production through an unknown mechanism in keratinocytes. In addition, α-toxin triggers a calcium mobilization response as well as TNF-α production through the formation of a heptameric transmembrane pore.
The currently available biologic agents for treatment of psoriasis have serious side effects as a result of immunomodulation. Therefore, many researchers have tried to develop new therapeutic agents for psoriasis that have a higher therapeutic activity, but fewer side effects. AMPs are a class of peptides synthesized by every type of living organisms to quickly eliminate Gram-positive and negative bacteria, fungi, parasites, enveloped viruses, and tumor cells,5051 and are promising as future therapeutic agents.5253 Natural AMPs are involved in host defense mechanisms in a variety of organisms such as cecropins in insects,54 Magainin in frogs,55 and defensins in rabbit, guinea pig, rat, and human neutrophil cells.56 AMP molecules are short (10–50 amino acids), with an overall positive charge and have both potent antimicrobial activity and anti-inflammatory effects. Recently, manipulation of natural peptides and synthetic peptides through modification of amino acids led to the development of a novel class of antimicrobial agents, which could be an alternative to the use of conventional antibiotics. As shown in Fig. 2, a direct interaction of S. aureus with keratinocytes or psoriatic keratinocytes plays a critical role in inflammation. Several reports have suggested that S. aureus promotes inflammation by stimulating keratinocytes to produce a number of proinflammatory cytokines such as VEGF, TNF-α, IL6 and IL-8 as well increasing expression and secretion of AMPs through TLRs-dependent pathways. The antimicrobial and anti-inflammatory activity of the synthetic AMPs may be attributed to its direct bactericidal activity and the ability of synthetic AMPs to bind bacterial factors such as peptidoglycan (PGN), lipoteichoic acid (LTA), protein A, α-Toxin, and superantigens (SAg). Synthetic AMPs have inhibitory activity on the secretion of proinflammatory cytokines such as tumor necrosis factor-α (TNF-α) and IL-8, chemokines, endogenous AMP, and VEGF in keratinocytes (Fig. 2). Thus, these properties make cationic AMPs attractive drug candidates for the treatment of psoriasis, a disease with both bacterial and inflammatory components.
Antimicrobial peptides and proteins act as moderators to link innate and adaptive immune mechanisms. Excessive production of AMPs induced by S. aureus is thought to modify the host inflammatory responses by a variety of mechanisms. They function as chemotactic agents, angiogenic factors, and regulators of cell proliferation. Recent advances highlight the role of the AMPs' cathelicidin, S100 proteins, and defensins in disease susceptibility and manifestation of psoriasis. These AMPs play a significant role in interactions between resident keratinocytes and skin infiltrating immune cells.57 Skin injury and infections such as S. aureus trigger rapid expression of human cathelicidin, hCAP18, in keratinocytes or infiltrated neutrophils. LL37 binds to extracellular self-DNA fragments, allowing them inside plasmacytoid dendritic cells (pDCs) to express type I interferons (IFNα and β) via TLR9 activation. As a consequence, the type I IFNs activate Th1 or Th17 cells through myeloid dendritic cells, which leads to the expression of INF-γ, IL-22 and IL-17.5859 IL-22 and IL-17 expression leads to more production and secretion of LL37, resulting in the pro-inflammatory feedback loop seen in psoriasis. AMPs are overexpressed in psoriatic lesions and function as alarmins on keratinocytes and other immune cells leading to heightened production of proinflammatory mediators and chemoattractant molecules for leukocytes. Thus, the normalization of aberrant, up-regulated expression of AMPs which is induced by S. aureus in keratinocytes and the inhibition of the early steps of inflammatory cascade in psoriasis might provide a therapeutic target in psoriasis. One problem in inhibiting AMPs is significant immunosuppression therefore this effect must be further investigated. While upregulation of AMPs worsen some aspects of psoriatic lesions, recent evidence has also suggested the possibility that the induction of AMP expression might improve certain aspects of psoriasis.42 Dombrowski et al.60 have discovered the anti-inflammatory effects of LL-37, which can block the activation of the DNA-sensing inflammasomes. This might be a mechanistic explanation regarding the effectiveness of vitamin D3 in psoriasis. It was discovered that intracellular or cytosolic LL-37 inhibits IL-1β release by blocking AIM2 inflammasomes in keratinocytes.60 Evidently, regulation of AMP expression and processing has an important role in psoriasis, but much remains to be learned about how they function.
2. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) in psoriasis
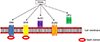
The tumor necrosis factor-related apoptosis-inducing ligand is a cytokine member of the tumor necrosis factor (TNF) family, which has potent cytotoxicity to tumor cells and transformed cells. TRAIL is a type II membrane protein that has an initial size of 33–35 kD, but can be cleaved from the cell surface by the aspartic proteinase cathepsin E to produce a soluble ligand of about 21 kD that retains biological activity. Once membrane bound, the extra-cellular domain of TRAIL assembles into a bell-shaped homo-trimer and three molecules of TRAIL fuse with three molecules of the transmembrane receptor as a hexameric complex (3:3). TRAIL binds to five distinct receptors found on a multitude of cell types; four of these receptors are membrane-bound and one a soluble receptor. Two of these membrane receptors, TRAIL-R1/death receptor 4 (DR4) and TRAIL-R2/death receptor 5 (DR5), function agonistically, and each incorporate a cytoplasmic death domain by which TRAIL propagates an apoptotic signal. On the contrast, the other two membrane receptors, TRAIL-R3/decoy receptor 1 (DcR1) and TRAIL-R4/decoy receptor 2 (DcR2), have affinity to bind TRAIL, but function as antagonistic/regulatory receptors without a death domain. Along with these four transmembrane receptors, an additional soluble, antagonistic receptor, osteoprotegerin (OPG), has been recognized (Fig. 3). TRAIL serves as a crucial regulator of immune responses during infections and autoimmune disorders. Its role has been investigated in skin cancers, various inflammatory diseases, and autoimmune-mediated pathologies. TRAIL has the dual ability of inhibiting angiogenesis by triggering endothelial cell death or enhancing pro-angiogenic activity in vitro.61
TRAIL has a strong ability to induce apoptosis in a concentration dependent manner. In fact, low concentrations of TRAIL can stimulate non-apoptotic action by promoting cell proliferation. Activated immune cells such as natural killer (NK) cells, T cells, macrophages and dendritic cells express membrane-bound TRAIL. On the contrast, soluble TRAIL is found in the sera of both normal individuals those affected by neoplastic disorders. Soluble TRAIL has also been detected in the culture supernatant of activated peripheral blood mononuclear cells (PBMC) when exposed to interferon. Therefore, it is thought to mediate antitumor cytotoxicity and function as an immune effector molecule. Interestingly, a significant level of TRAIL transcript has been discovered in many human tissues such as the colon, ovary, PBMC, placenta, prostate, spleen, small intestine, and thymus. In addition, TRAIL has constitutive expression in some tissues. In order to best characterize the biological activity of both full-length membrane and soluble TRAIL, Multiple experimental in vitro and in vivo models of the induction of apoptosis in neoplastic cells by TRAIL have been developed . Several studies have demonstrated that the TRAIL-TRAIL receptors system play a physiological role in normal angiogenesis through promotion of endothelial cell survival, migration, and proliferation. An anti-inflammatory activity of TRAIL has also been demonstrated by observing how TRAIL responds to inflammatory cytokines by counteracting the adhesion of both peripheral blood derived monocytes and granulocytes to endothelial cells in vivo without activating apoptosis.61
TRAIL has been hypothesized to play a role in the pathogenesis of psoriasis. Caldarola et al.62 investigated the expression and localization of TRAIL, its receptors, and the intracellular pathways involved with TRAIL to elucidate any involvement in psoriasis pathogenesis. They reported increased production of TRAIL and its receptors in both lesional vs. non-lesional skin using RT-PCR. Also, TRAIL and TRAIL receptors were found in the epidermis and in the dermal infiltrate by immunohistochemistry. Finally, they found that caspase 8 and TRAIL expression in the dermis were correlated, hypothesizing that caspase 8 action at the site of the inflammation may explain these phenomena.62
In psoriatic lesions, IFN-γ was observed to stimulate IL-23 and IL-1 production by DCs which ultimately activated Th17 cells in the inflammatory cascade. IFN-γ was intradermally injected once to a clinically normal and nonlesional area of skin in psoriasis patients and biopsies were collected after 24 hours. Although no visible changes were observed in the skin, areas injected with IFN-γ revealed many molecular and histological features typical of psoriatic lesions. IFN-γ also upregulated a number of differentially expressed genes in the skin as well as chemokines responsible for an influx of T cells and inflammatory myeloid DCs. Furthermore, the IFN-γ-treated skin contained inflammatory DC products tumor necrosis factor (TNF), inducible nitric oxide synthase, IL-23, and TRAIL, which further supports involvement of TRAIL in psoriasis pathogenesis.63
The expression of TRAIL and its receptors DR4 and DR5 has been investigated in psoriasis vulgaris through immunohistochemistry staining. Notably, TRAIL stains were significantly pronounced in both psoriatic lesional and non-lesional epidermis when paralleled with the epidermis of healthy skin. There was also increased immunoreactivity for DR5 in lesional epidermis. Additionally, expression of TRAIL and both of its receptors was considerably amplified in the dermis of lesional skin. Cells of the inflammatory infiltrate in psoriatic lesions abundantly expressed TRAIL. The expression of DR4 was found localized in the nucleus of CD4+ and CD8+ cells, while in contrast, DR5 demonstrated mostly cytoplasmic staining in rare CD16+, CD56+ and CD68+ cells. Considering the abundant in situ presence of TRAIL and its receptors found in lesional psoriatic skin, there appears to be a complex relationship between keratinocytes and cells of the dermal infiltrate in psoriasis vulgaris.64
Zaba et al.65 previously reported that CD11c(+)CD1c(−) inflammatory dermal dendritic cells (DCs) in psoriatic lesional skin secrete a wide variety of inflammatory molecules compared with skin-resident CD1c(+) DCs. The differential expression of TRAIL, TLR1, TLR2, S100A12/ENRAGE, and CD32 were shown in inflammatory DCs compared to resident DCs. Keratinocytes treated with TRAIL expressed TRAIL receptors, death receptor 4, and decoy receptor 2; in contrast, dermal cells during in vitro culture of keratinocytes with TRAIL induced CCL20 chemokines. Considering these results, molecular products made by inflammatory DCs, including TRAIL as discussed above, have the potential to modulate keratinocytes or other skin cell types in psoriasis pathogenesis.65
It has recently been reported that TRAIL inhibits VEGF-induced angiogenesis of human umbilical vein endothelial cells (HUVECs) by increasing caspase-8 activity. TRAIL also decreases procaspase-8 associated non-apoptotic signaling functions without inducing caspase-3 activation and endothelial cell cytotoxicity. Since VEGF is an important factor in the development of psoriasis and psoriasiform like plaques in VEGF-transgenic mice, caspase-8 is a promising target for an anti-angiogenic drug for psoriasis.66
It has been reported that a novel modification mechanism of TRAIL signaling by receptor-interacting protein kinase 1 (PIPK1) in epidermal keratinocytes may offer a novel therapeutic target in psoriasis.67 RIPK1, a death domain-containing serine/threonine protein kinase, acts as regulator of apoptosis and necroptosis downstream of TNF-α, TRAIL, and TLRs, and skin inflammation.68 Recent studies have shown that keratinocytes in psoriatic lesions are highly sensitive to TRAIL signaling by RIPK1-downregulation.67 The 80 to 90% decreased expression of RIPK1 was reported in psoriasiform skin compared with that of a healthy control of IMQ-treated mice, an animal model recapitulating key features of psoriasis.69 The psoriasis-relating cytokines such as IL-1β, IL-17α, IL-22, TRAIL, and IL-17α plus IL-22 significantly reduced RIPK1-expression in human epidermal keratinocytes (HEK). This allows TRAIL to induce psoriasis-related cytokines such as IL-1β, IL-6, IL-8, IL-20, IL-33, and TNF-α via the NF-κB pathway in RIPK1-decreased keratinocytes. Finally, keratinocytes increased their TRAIL susceptibility through RIPK1 downregulation in the absence of any type of cells.67
Since LL-37 highly upregulated TRAIL receptor expression in psoriasis-relating cytokines-induced human epidermal keratinocytes (HEK), and since a complex of LL-37 and host DNA stimulated dermal plasmacytoid DCs to produce IFN-α at the psoriasis onset and during the exacerbation stage, the TRAIL upregulation via LL-37 may be implicated in the initial stages of psoriasis.67 Numerous TRAIL-positive cells in the epidermis of IMQ-model mice strongly expressed TRAIL receptor and DR5 compared with that in the control. In contrast, the expression of RIPK1 in the epidermis was decreased mirroring the decrease in psoriatic epidermis. Neutralization of TRAIL suppressed the skin phenotype of IMQ-treated mice with a marked decrease in erythema, infiltration, and scales as well as histopathological improvement of acanthosis, hyperkeratosis, and parakeratosis. These preliminary results clearly support future research on new approaches to psoriasis treatment using modulation of RIPK-1 signaling and LL-37.
3. Neuropilin-1 (NRP1) blocking peptides
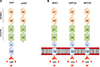
Neuropilin-1 (NRP1), a type 1 transmembrane protein, is expressed in neurons, blood vessels, immune cells, and many other cell types in mammals. NRP1 has been reported to play a role in the attenuation of autoreactive immune responses as well as regulation of angiogenesis and permeability induced by VEGF, ECM and semaphoring-3A.707172 As shown in Fig. 4A, there are two types of NRP1: a membrane-bound full-length form and two soluble forms. Full length NRP1 consists of seven domains - two complement (CUB) domains (a1 and a2), two coagulation factor (FV/FVIII) domains (b1 and b2), a MAM domain (c), a transmembrane region (TM) and a cytoplasmic domain (cyto). The NRP1 cytoplasmic domain has known to interact with the PDZ domain contained-intracellular proteins. Since all soluble NRP1 isoforms lack the c, transmembrane and cytoplasmic domains (Fig. 4A), they are not able to transduce signals, resulting in their function as decoy receptors to sequester NRP1 ligands.73 Human NRP1 and two NRP2 alternative splicing variants, such as NRP2A and NRP2B, have high similarities with the amino acid sequences of the corresponding a, b, and c domains, which are 45%, 48%, and 35% identical as shown in Fig. 4B.727475 NRP1 has 53% similarity in the cytoplasmic domain with NRP2A and also has a SEA motif. In contrast, NRP2B is not able to interact with PDZ domain-contained proteins due to lack of the SEA motif in its cytoplasmic domain.76
Class 3 semaphorin A (SEMA3) regulates axon guidance as a secreted glycoprotein7778 and VEGF A receptor.7980 Recent studies reported that NRP1 is a SEMA3 receptor, and NRP2 is a VEGF isoform receptor in the SEMA3 family. Furthermore, NRP1 and NRP2 interact with integrins in various cell types.8182838485
Many studies have shown an involvement of NRP1 in psoriasis pathogenesis. Recently, these studies have shown that NRP1 expression was increased in psoriatic skin.86 NRP1 expression is related to the proliferation or differentiation program of keratinocytes and is expressed in the basal and supra basal layers of the skin epidermis. It is also expressed in fibroblasts, endothelial cells, and inflammatory cells of the dermis. However, the most intensive expression of NRP1 was observed in psoriatic skin. Highly elevated expression of NRP1 mRNA was detected in the epidermis and the dermis of psoriatic skin when compared with healthy skin. In addition, NRP1 expression is closely correlated with the expression of Ki67, a well-established proliferation marker. Moreover, it was reported that the signaling pathway of NK-κB is one that is putatively downstream of NRP1-associated signaling in HaCaT cells. This study suggests that NRP1 involvement in regulation of proliferation is through activation of the NF-κB pathway in keratinocytes in psoriatic skin. Furthermore, the expression of angiogenesis markers including soluble 12 NRP-1, VEGFR3 and VEGF-A isoform VEGF121 was highly increased in the non-lesional psoriatic skin as compared with that of normal healthy skin.8687 Saban et al.88 reported that NRPs and VEGFRs play a role in migration of inflammatory cells and inflammation-induced lymph-angiogenesis. In this study, neutralizing antibodies against NRPs blocked the binding of VEGF to NRP (e.g. anti-NRP1 (B)) and the binding of semaphorins to NRP (anti-NRP1 (A)) in the BCG challenged C57BL/6 mice. They indicated that NRP1-neutralizing antibodies dramatically reduced scVEGF/Cy5.5 uptake as well as polymorphonuclear (myeloperoxidase+cells) and dendritic cell (CD11c+positive cells) infiltration in the NRP1-treated mice. The NRP1 treatment also decreased the overall density of BCG-induced blood and lymphatic vessels, indicating that NRPs play a critical role as in vivo regulators of the vascular and inflammatory responses in the BCG challenged C57BL/6 mice.88 Since vascular alterations are a significant hallmark in the pathogenesis of psoriasis, angiogenesis is a central process in the evolution of psoriasis. As such, anti-angiogenic treatments of psoriasis remain under evaluation for effectiveness. Therefore, targeting NRP1 interaction with VEGF/VEGFR, semaphorins may be a significant approach for intervention in psoriasis progression.
CONCLUSION
Although there are numerous effective treatments for psoriasis that target specific points in the pathogenic pathways that control psoriasis, there is no cure for psoriasis currently. To date, the pathogenic processes that control inflammatory skin disease are clinically characterized by prominent epidermal hyperplasia and a distinctive inflammatory infiltrate. Although psoriasis cannot be considered uniquely as a T-cell-dependent disease, the initiation, progression, and persistence of psoriasis is influenced by inflammation and immune circuits generated by the cross talk between autoreactive T cells and resident keratinocytes in psoriatic skin. As shown in Fig. 1, the disease chronicity in psoriasis may be determined by aberrant interplay of immune cells and cutaneous keratinocytes in intensified inflammation and autoimmune responses. Since keratinocytes also play a critical role in innate and adaptive immune responses, keratinocyte activation is characterized by the over-proliferation and overexpression of immunological molecules that are critical mediators in the development of psoriasis.
Angiogenesis is another central process in the development of psoriasis. For this reason, potential intervention on angiogenesis and keratinocyte activation represents an attractive therapeutic target against psoriasis. In this review, we focused on current knowledge about the emerging role of synthetic AMPs, TRAIL and NRP1 blocking peptides on angiogenesis and keratinocyte activation for the pathogenesis and treatment of psoriasis.
Considerable evidence suggests an important role for bacteria such as S. aureus, tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), and neuropilin 1 (NRP1) in the pathogenesis of psoriasis. Although psoriasis is classified as an autoimmune disease, it is now well accepted that colonization and/or infection with the bacteria such as Staphylococcus and Streptococcus in the skin have an important role in the pathogenesis of this condition. A promising approach for developing new cutaneous, anti-bacterial agents may lie with antimicrobial peptides because of their ability to efficiently kill microbial pathogens in addition to their anti-inflammatory effects. In this review, we have discussed TRAIL and its receptors DR4 and DR5, which are strongly expressed in plaque psoriasis. In addition, TRAIL has the ability to inhibit angiogenesis by inducing endothelial cell death and negatively regulates VEGF-induced angiogenesis via caspase-8-mediated enzymatic and non-enzymatic functions. We also emphasized NRP1 as a co-receptor for VEGF and its role in regulating angiogenesis through signaling pathways associated with VEGF, ECM, and semaphorins. Given this background, novel modifications of synthetic AMPs, TRAIL, and NRP1-signaling in epidermal keratinocytes and angiogenesis are a potential target for psoriasis therapies. Further studies are clearly needed to define the specific mechanisms by which these molecules regulate angiogenesis and keratinocyte activation in order to better understand the pathogenesis and treatment of psoriasis.
 XML Download
XML Download