

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
The prognosis after acute myocardial infarction (AMI) has improved with advances in percutaneous coronary intervention (PCI) and pharmacologic therapies.1 However, the risk of recurrent major adverse cardiovascular events (MACE) beyond the first year post-AMI remains high.2 Compared with stable coronary artery disease (SCAD) patients, AMI patients show worse clinical outcomes in deferred lesions based on non-ischemic fractional flow reserve (FFR) despite optimal medical therapy34 as well as more frequent stent restenosis and thrombosis after PCI.56
The presence of non-culprit lesions in AMI is associated with increasing 30-day mortality and MACE similar to the culprit lesion.78 Recent studies primarily focused on finding high-risk atherosclerotic plaque features to predict events,89 and performing preventive PCI with FFR or angiography guidance to improve clinical outcomes.10111213 Local plaque vulnerability alone, however, cannot fully explain future events after AMI such as cardiac death and recurrent infarction.141516 AMI triggered a burst of acute systemic inflammation and activation of hematopoietic organs such as the bone marrow and spleen.1718 There have been several reports of a murine AMI model in which an inflammatory reaction could be associated with myocardial repair, pre-existing plaque progression, and heart failure.192021 Recent study showed that AMI promoted pro-inflammatory endothelial activation in remote arteries such as the thoracic murine aorta.22
Here we aimed to evaluate the influence of local AMI events on endothelial function, neointimal progression, and inflammation in target and non-target vascular territories in a porcine model of closed-chest ischemia-reperfusion AMI.
METHODS
Animal preparation
The study included castrated male pigs weighing 20–25 kg each. To decrease the risk of cardiac death after the induction of AMI, premedication with aspirin (100 mg/day) and clopidogrel (75 mg/day) was administered 5 days before the procedure. Preparation of in vivo experiment prior to AMI induction and stent implantation was reported previously.23 Briefly, the guiding catheter is placed in the left coronary artery through the left carotid artery after general anesthesia.2324
Induction of AMI
Coronary vasoreactivity was examined by an acetylcholine infusion before wiring. The procedure was only continued in animals without paradoxical coronary vasoconstriction. The AMI was induced by a 15-mm balloon at the distal portion of the first diagonal branch or the septal branch of the left anterior descending artery (LAD). The diameter of the balloon was adjusted to the vessel size with reference to the 7-Fr guiding catheter diameter (2.31 mm). Coronary occlusion was maintained by balloon dilatation for 60 minutes.25 Oxygen and normal saline were supplied continuously, and the anesthesia was maintained with inhalation (isoflurane 1%) during the experiment. Continuous electrocardiographic monitoring was performed to confirm a normal ST segment at baseline and an ST elevation during the ischemic period and monitor for the occurrence of cardiac arrhythmia. After the induction of AMI, each pig was closely observed for 3 hours for the development of ventricular tachycardia or fibrillation. Coronary angiograms were obtained immediately after balloon deflation and intracoronary nitroglycerine bolus injection to confirm antegrade coronary flow.
Sham operation and complete blood cell count analysis
Sympathetic nerve activation and inflammation could affect the present study results. To minimize intergroup differences, the control group was subjected to cut-down, guiding engagement, coronary angiography, wiring, and balloon passage similar to the AMI group. General anesthesia persisted for 4 hours. Complete blood cell count analysis (HD-URIT-3000 Vet Plus Automatic Hematology Analyzer, Shanghai, China) was performed to determine whether severe anemia or severe inflammation had developed peri-procedurally. Blood sampling was done at three time points (before and 2 days after AMI induction and the day the pigs were sacrificed) in the two groups.
Implantation of drug-eluting stents
Two days after the AMI and sham operations, the pigs were anesthetized as follows. The left circumflex (LCX) stents (non-target vessel) were deployed by inflation of the balloon to achieve a stent-to-artery ratio of 1.3:1 at the normal coronary artery using an everolimus-eluting stent (EES; Xience Xpedition®; Abbott Vascular, Santa Clara, CA, USA). The diameter of the implanted coronary stent (stent-to-artery ratio) was adjusted with reference to the 7-F guiding catheter diameter. The LAD stents (target vessel) were implanted below the balloon-injured lesion. Four weeks after stenting, the animals underwent follow-up angiography from the same orthogonal views before being euthanized with an intracoronary injection of 20 mL of potassium chloride. The heart was removed and the stented coronary arteries were pressure-perfusion fixed at 110 mmHg overnight in 10% neutral buffered formalin solution. Each group of 10 stented arteries was step-sectioned, processed routinely for light microscopy examination, and stained for comparative histological analysis. Each group of 10 stented arteries was step-sectioned, processed routinely for light microscopy examination, and stained for comparative histological analysis.
Endothelial function assessment
Assessment of coronary endothelial function was reported previously.23 Acetylcholine was injected into the guiding catheter (50 µg and 100 µg for 1 minute; infusion rate, 5 mL/min) before wiring (before AMI), before stenting (2 days after AMI), and at 1-month follow-up.
Histopathological analysis of stented artery
The histopathological evaluation of each artery was performed by an experienced cardiovascular pathologist. Histopathological analyses were performed in a blinded fashion. Detailed methods were reported previously.23 Degree of injury, inflammation, fibrin contents were graded according to the former studies.262728
Statistical analysis
All analyses were performed using Statistical Package for the Social Sciences software version 21.0 (IBM, Chicago, IL, USA). Continuous variables are presented as mean ± standard deviation, and comparisons were made by Mann-Whitney non-parametric tests. The χ2 test or Fisher's exact test was used to determine the significance of the differences between categorical variables. Pearson's r coefficient of correlation was used for the correlation studies. All probability values were two-sided and P values < 0.05 were considered statistically significant.
Ethics statement
The present animal study was approved by the Ethics Committee of Chonnam National University Medical School and Chonnam National University Hospital (CNU IACUC-H-2010-18) and conformed to Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996).
RESULTS
Baseline status of experimental animals
A total of 14 pigs were prepared for this study. Of them, two were excluded before wiring due to paradoxical vasoconstriction after the acetylcholine infusion. In three pigs, ventricular fibrillation occurred during AMI induction, which was terminated by cardiac defibrillation and amiodarone infusion in the peri-procedural period. However, one died 1 day post-AMI. Before stenting (2 days post-AMI), one pig showed total LAD occlusion and ventricular fibrillation after the acetylcholine infusion that were not restored despite repetitive intra-coronary nitroglycerine infusion, cardioversion, and epinephrine and amiodarone injections. Finally, 10 pigs were implanted with a total of 20 EES in the LAD and LCX. No additional deaths occurred during the 1-month follow-up period. Fig. 1 summarizes the protocols of the present study.
 | Fig. 1Study Protocol. To minimize intergroup differences, the control group was subjected to cut-down, guiding engagement, coronary angiography, wiring, and balloon passage similar to the AMI group. Endothelial function was assessed by measuring the coronary vasomotor responses to incremental doses of acetylcholine into the guiding catheter (50 µg and 100 µg for 1 minute; infusion rate, 5 mL/min) before wiring (before AMI), before stenting (2 days after AMI), and at 1-month follow-up.AMI = acute myocardial infarction, DES = drug eluting stent, CAG = coronary angiogram, FU=follow up, CBC = complete blood cell count, Ach = acetylcholine, ASA = aspirin.
|
Vasomotor responses post-AMI and -PCI
Mean stent diameter (control 2.7 ± 0.2 mm vs. AMI 2.7 ± 0.2 mm; P = 1.0), stent length (control 15.0 ± 1.41 mm vs. AMI 15.3 ± 1.70 mm; P = 0.673), and stent deployment pressure (control 10.1 ± 1.29 mmHg vs. AMI 10.0 ± 1.70 mmHg; P = 0.870) were similar in the two groups. Compared with the control group, significant vasoconstriction occurred in both target (LAD, 44.7% ± 10.75%) and non-target (LCX, 17.9% ± 6.70%) vessels immediately post-AMI induction. Two days after the AMI and sham operations, a vasoreactivity test was performed before the EES implantation. Significant paradoxical vasoconstriction occurred after the acetylcholine challenge in the AMI group (−54.9% ± 33.87% vs. −5.2% ± 4.6%; P = 0.001) compared with the control group. At 1 month after the index procedure, both groups showed vasoconstriction after acetylcholine infusion. However, the AMI group showed more vasoconstriction than the control group (AMI −57.4% ± 15.62% vs. control −27.6% ± 9.10%; P < 0.001) (Supplementary Table 1). Compared with that of the control group, non-target vessel of AMI group also showed significantly high degree of vasoconstriction after acetylcholine infusion (AMI −47.0% ± 12.5% vs. control −27.7% ± 9.19% P = 0.026) (Table 1 and Fig. 2).
Table 1
Vasomotor responses in non-target vessel (LCX) between control group and AMI group

Data are presented as mean ± standard deviation.
LCX = left circumflex artery, AMI = acute myocardial infarction, Ach = acetylcholine.
![]()
 | Fig. 2Vasomotor responses post-AMI and -PCI. (A) Vasomotor responses 2 days after AMI induction between AMI and control group, (B) Vasomotor responses 1 month after PCI between AMI and control group, (C, D) Divide AMI group into target vessel (balloon inflation, LAD) and non-target vessel (not balloon inflation, LCX). (C) Vasomotor responses 2 days after AMI induction. (D) Vasomotor responses 1 month after PCI.AMI = acute myocardial infarction, PCI = percutaneous coronary intervention, LAD = left anterior descending, LCX = left circumflex.
*P < 0.05 versus control group; **P < 0.05 versus non-target vessel.
|
Histopathologic analysis on stented artery and myocardium
The AMI group showed a greater mean neointimal area than the control group after EES implantation in both target (1.5 ± 0.3 vs. 0.9 ± 0.2; P < 0.001) and non-target vessels (1.2 ± 0.4 vs. 0.9 ± 0.2; P < 0.05). Significant peri-strut inflammation and fibrin formation were found in the AMI group without differences in injury score, which could have affected the degree of neointimal hyperplasia (Table 2, Supplementary Table 2, Figs. 3 and 4).
Table 2
Histo-morphometric measurements according to the target and non-target vessel

AMI = acute myocardial infarction, SD = standard deviation, IQR = interquartile range, IEL = internal elastic lamina, LCX = left circumflex artery.
![]()
 | Fig. 3Histology of stented arteries. Representative images of hematoxylin and eosin staining at four weeks after stenting. Specimens of (A) implanted control group (magnification, ×20) and (B) AMI group (magnification, ×20). Carstair fibrin staining of the low power fields (magnification, ×20) in (C) implanted control group and (D) AMI group.AMI = acute myocardial infarction.
|
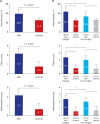 | Fig. 4Histopathological analysis. Percent area stenosis, fibrin score, and inflammation score for (A) AMI vs. control group and (B) target vs. non-target vs. control group.LAD = left anterior descending, AMI = acute myocardial infarction, LCX = left circumflex.
*P < 0.05.
|
The myocardium in the target and non-target territories of the AMI group was analyzed and consisted of five porcine hearts with a 1-month follow-up as well as two porcine hearts at 1 and 2 days after AMI induction. Inflammatory changes and myocardial necrosis were observed in the target vessel territory myocardium and microvasculature such as the arterioles but not in the non-target vessel territory (Fig. 5).
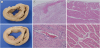 | Fig. 5Pathologic analysis in myocardium and microvasculature. (A) Wall thinning and whitish changes are noted in the anterior wall and anterior portion of the interventricular septum on the macroscopic examination. (B) Massive fibrosis, chronic inflammation, and capillary proliferation are noted in the target vessel territory on the microscopic examination (H&E stain ×40). Both periarteriolar (*) and perivenular (**) inflammation are noted in the fibrotic lesion on the microscopic examination (H&E, ×200). (C) There are no specific findings in the non-target vessel territory myocardium and microvasculature on the microscopic examination (H&E stain ×40, ×200).H&E = hematoxylin and eosin.
|
Complete blood cell count analysis
The baseline white blood cell count, hemoglobin level, and platelet count prior to the procedure were not significantly different between the two groups (Table 3). Compared with the control group, the white blood cell count was significantly elevated after AMI induction and returned to similar levels after 1 month of follow-up. No significant bleeding events or thrombocytopenia occurred after anti-platelet agent administration and the cut-down procedure.
Table 3
Hematological parameter values

![]()
DISCUSSION
In this investigation, we explored the influence of AMI in the LAD on endothelial function, neointimal progression, and inflammation in target and remote vessels using a porcine AMI model. The main results are as follows. Firstly, the non-target vessel of AMI also showed similar findings as the target vessel compared with the control group. Secondly, significant endothelial dysfunction developed after AMI induction during the 1-month follow-up. However, necrotic and inflammatory changes were not observed in the non-target vessel myocardium and microvasculature. Thirdly, post-AMI stenting was associated with more neointimal progression, peri-strut inflammation, and fibrin formation.
Influence of AMI on non-target vessel
Endothelial dysfunction assessed by intracoronary acetylcholine during coronary angiography was associated with plaque progression, stroke, and AMI in patients with mild coronary artery disease.293031 Several data suggest that endothelial dysfunction could be developed due to AMI itself in culprit and non-culprit lesion. First, AMI triggers endothelial activation in remote vessels including the up-regulation of adhesion molecules of inflammatory cells, endothelial-associated von Willebrand factor, and secondary platelet adhesions. However, this murine study lacked an endothelial function test.22 Second, coronary flow reserve (CFR) was depressed due to an increasing resting flow velocity and decreasing hyperemic flow velocity in culprit and non-culprit lesions in AMI patients compared with SCAD patients.323334 These studies, however, could not prove whether AMI lowered CFR or AMI occurred more in patients with lowered CFR.35 Serial measured data rather than matched patients' data were needed to clarify this issue. However, it is difficult to obtain baseline human data before AMI. Third, endothelial dysfunction was observed in about 81% of non-culprit and angiographically normal coronary arteries of patients with non-ST segment elevation myocardial infarction. We could not determine the presence of underlying endothelial dysfunction, and the application in patients with more unstable AMI (e.g., ST-segment elevation myocardial infarction). In fact, endothelial function testing during AMI might be limited due to a patient's unstable condition and dramatic impairment in microvascular function.36 In the present study, we showed that significant endothelial dysfunction developed simultaneously after AMI induction in target and non-target vessel lesions. This global endothelial dysfunction after AMI was demonstrated to be associated with MACE in a previous study.37
Drug-eluting stent implantation in AMI had a higher frequency of stent malapposition, delayed tissue coverage, plaque protrusion, and smaller minimal stent area.63839 These studies primarily focused on the relationship between vulnerable plaques and stent struts only in the culprit lesion of the AMI. Because the AMI could occur in the non-obstructive lesion or the plaque erosion,4041 these findings did not fully explain why PCI in AMI was associated with a poor prognosis. The present study showed that AMI itself was associated with stent restenosis, peri-strut inflammation, and fibrin formation, findings that were simultaneously observed in the non-target vessel of the AMI. As there was no preexisting plaque in the present animal study, neointimal formation might be associated with peri-strut inflammation and endothelial dysfunction.2542
Coronary epicardial endothelial dysfunction and microvascular resistance
AMI in the target vessels results in global endothelial dysfunction of the non-target vessels. We previously performed a porcine experiment to explore the influence of microvascular damage in the target vessel territory to changes in the index of microvascular resistance (IMR) in the non-target vessel territory.43 Microvascular dysfunction developed gradually using five microsphere injections into the LAD. IMR values were elevated in the LAD but not significantly changed in the LCX. Ntalianis et al.44 study showed that IMR values in the non-culprit lesion were similar at the acute phase and 1 month follow-up and that about 80% of the IMR values measured in non-culprit vessels were within the normal range. Recent data also showed that non-culprit IMR values did not differ between the in 100 AMI and matched SCAD patients.34 In the present study, myocardial injury and inflammatory reactions within the microvasculature were not observed differently in the non-target vessel territory after AMI induction as compared to the target-vessel territory. The present study, however, lacked serial physiologic measurements in CFR and IMR; it is, thus, difficult to make definite conclusions about the relationship between coronary epicardial endothelial dysfunction and microvascular resistance.
Clinical implications
The present study was conducted to assess why AMI non-culprit lesions have poor clinical outcomes similar to culprit lesions. Pre-existing plaque vulnerability in non-culprit lesions was considered as a major determinant of MACE in the Providing Regional Observations to Study Predictors of Events in the Coronary Tree (PROSPECT) and VH-IVUS in Vulnerable Atherosclerosis (VIVA) study.89 However, there are several reports that plaque vulnerability solely cannot fully explain future events after AMI such as cardiac death and recurrent infarction.141516 Endothelial dysfunction and inflammation could be the triggering factors for MACE in AMI non-culprit lesions. However, it is difficult to distinguish the cause from effect between these factors and AMI in a human study.
In this swine model, AMI events induced endothelial dysfunction, inflammation, and neointimal progression in the target and non-target vessels. Systemic burst inflammatory reaction was developed after AMI in acute stage (around 2 days), and relieved in chronic stage (around 1 month); significant endothelial dysfunction was observed in both periods after AMI. These findings support the facts that AMI patients have worse clinical outcomes than those with stable coronary artery disease despite revascularization, and non-culprit lesions also involve more adverse cardiovascular events regardless of underlying plaque vulnerability.
Therefore, the treatment of AMI should not simply concentrate on reducing the vulnerable plaque burden by PCI. The duration of dual antiplatelet therapy (DAPT) including aspirin plus P2Y12 inhibitors gradually decreased as drug-eluting stents improved in patients with SCAD, but not in AMI.45 Unlike SCAD, the discontinuation of DAPT within 1 year might be associated with MACE.4647 A prior AMI could be a substrate of future ischemic events beyond 1 year,248 and the longer-term (beyond 1 year) use of the potent P2Y12 inhibitor ticagrelor significantly reduced the risk of cardiovascular death, AMI, or stroke for a median 33 months of follow-up.49 Reducing the inflammation reflected by high-sensitivity C-reactive protein improved outcomes in patients with AMI regardless of the low-density lipoprotein cholesterol reduction.5051 The recent Canakinumab Anti-Inflammatory Thrombosis Outcomes Study showed the possibility of treating residual inflammatory risk in prior AMI patients using the anti-inflammatory agent.52 However, there are still long way to go to find effective, inexpensive, widely used anti-inflammatory agents for preventing atherosclerotic events.53
Limitations
This study has some limitations. First, no previous study serially measured endothelial function, neointimal area, or inflammation score after the induction of AMI. Therefore, it was not possible to assume or calculate sample sizes before the present experiment. We decided to use a generally accepted size (10 vessels in each group) to compare neointimal area and inflammation score as in previous porcine studies. The results were consistent and already statistically significant; we did no additional experiments on the basis of the 3R code (replacement, reduction, refinement) of animal experiments. Second, AMI induced by balloon occlusion and reperfusion may not fully reflect the complex pathophysiology of myocardial damage in the culprit vessels of AMI patients. Third, this study lacks the mechanistic explanations for the present findings using the analysis of target genes, signal pathways, and proteins.
 XML Download
XML Download