

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Antiphospholipid syndrome (APS) is a multisystem autoimmune disease characterized by arterial thrombosis, venous thrombosis associated with persistently positive antiphospholipid antibodies (aPL) [1]. aPL comprises three main antibodies: lupus anticoagulant (LA), anticardiolipin, and anti-beta2-glycoprotein I (anti-β2-GPI). APS may occur alone or in conjunction with systemic lupus erythematosus (SLE) or rheumatoid arthritis (RA) [2]. However, the pathophysiological mechanisms of this correlation are unclear [3].
Catastrophic APS is a rare, severe, and life-threatening form of APS resulting in multiorgan failure and affects approximately less than 1% of patients with APS [4]. Furthermore, the low prevalence of pediatric catastrophic APS makes its diagnosis difficult. Therefore, early diagnosis and aggressive treatments are very important. The preliminary criteria for the classification of catastrophic APS were presented by the International Task Force in 2003 [4], and were ultimately validated by an extended report published in 2005 [5]. These criteria help clinicians recognize circulating aPL and the rapid onset of thrombosis in multiple organs (Table 1) [5].
Because of currently available conventional combined treatment regimens, such as anticoagulants, corticosteroids, plasmapheresis, and immunoglobulins (IVIGs), the mortality rates have decreased from 53% to 37%, compared before 2001 [16]. Despite the significant decrease, the mortality rate is still high and there is no definite effective treatment. Recently, rituximab (Mabthera) has been shown to be effective as a treatment for catastrophic APS. However, there are few studies on the use of rituximab in treating catastrophic APS in both adults and children.
We herein report the case of a 14-year-old girl with catastrophic APS who was successfully treated with rituximab.
CASE REPORT
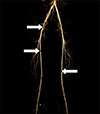
A 14-year-old girl was admitted with right fifth toe's pain and necrotic changes. She was diagnosed with SLE at the age of 11 and was continuously treated with hydroxychloroquine, prednisolone (PD) and azathioprine. At that time, she was in the process of tapering the dose of PD, and was taking it at a dose of 5 mg twice a day, just before admission. Three months before admission, she experienced acute pain with skin color change in right fifth toe, so she received with aspirin treatment (100 mg daily) additionally. At the time of admission, she had no fever and her vital signs including blood pressure (BP) were stable. Laboratory results showed hemolytic anemia (hemoglobin [Hb] 6.6 g/dL), positive Direct/indirect Coombs' test, prolonged activated partial thromboplastin time (aPTT) (99.5 seconds), increased total bilirubin (2.38 mg/dL) (particularly indirect bilirubin 1.79 mg/dL), and disseminated intravascular coagulation (DIC) profiles such as fibrin degradation product (FDP), D-dimer, fibrinogen levels were within normal range. She tested positive for anti-dsDNA, anti-nuclear antibodies, anticardiolipin IgM and IgG antibodies, anti-β2-GPI and LA. Ultrasonography (US) and computed tomography (CT) angiography of the lower extremity showed multiple stenoses in the right distal common iliac artery, right upper superficial femoral artery, and left lower superficial femoral artery (Figure 1). Despite the treatment, the symptoms did not resolve, and necrotic changes gradually worsened on the right fifth toe. Therefore, percutaneous transluminal angioplasty was performed, and low-molecular- weight heparin (LMWH) was added to the medication. However, autoamputation of the right fifth toe occurred six months later.
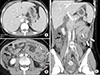
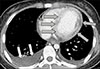
Three months later, she was again admitted for vomiting, diarrhea, and abdominal pain without fever. On initial examination, her vital signs were stable and there were no clinical signs of acute peritonitis. Laboratory results showed hemolytic anemia (Hb 5.6 g/dL), thrombocytopenia (platelet count, 40×103/µL), elevated C-reactive protein (93.4 mg/L [normal, 0.0~5.0 mg/dL]), and prolonged aPTT (99.5 seconds). Fibrinogen (683 mg/dL), FDP (7.91 µg/mL), D-dimer (1.93 µg/mL), and antithrombin III (128%) were elevated. She tested positive for LA, anticardiolipin IgM and IgG antibodies, and anti-β2-GPI. Abdominal US and CT revealed hepatosplenomegaly, long and segmental jejunal wall thickening with a large amount of ascites (Figure 2A~C). Thus, intravenous (IV) methylprednisolone (MPD) pulse therapy (1 g/day for 3 days) was initiated under the diagnosis of lupus enteritis. The day after initiation of MPD pulse therapy, shortness of breath, palpitation, and retrosternal pain suddenly occurred. On electrocardiography, ST-segment elevation was noted at lead II, III, and aVF, and 0.6-mg sublingual nitroglycerin was administered (Figure 3). Emergency laboratory tests showed elevated cardiac enzyme levels (creatine kinase [CK], 1,291 IU/L; CK-MB, 170.9 ng/mL; and troponin I, 16.15 ng/mL). Due to acute myocardial infarction (MI), emergency coronary angiography was performed, and minimal lesions were observed in the left circumflex coronary artery and the left anterior descending artery. On chest CT, low-attenuation lesions considered as ischemic changes around the left ventricle were observed with a large amount of pleural fluid (Figure 4). An Echocardiogram showed hypofunction of the right ventricle, and myocarditis was strongly suspected. On the eighth day of admission, she complained of chest and back pain with fever (body temperature of ≥38.5℃) and was identified as having reduced BP 80/40 mmHg. Thus, we administered inotropics, such as dopamine and dobutamine with analgesics. Thereafter, her symptoms and vital signs gradually improved, and MPD pulse therapy was restarted. With treatment, her symptoms subsided and platelet count also normalized.
One month later, she was admitted for the third time; this time, the presented with petechial lesions of the limbs and oral mucosal bleeding. Laboratory tests showed thrombocytopenia (platelet count, 6×103/µL) with prolonged aPTT (99.3 seconds) and DIC profiles were within normal range. The clinical course did not improve despite IVIG administration for 5 days at a dose of 2 g/kg once a day and transfusions. Thus, seven rounds of plasmapheresis were administered. After plasmapheresis, the platelet count increased to 57×103/µL; however, the effect was transient. Despite plasmapheresis, her condition showed no resolution, and ultimately, we decided to administer rituximab. Rituximab was administered at a dose of 375 mg/m2 by IV infusion once weekly for 4 weeks. The skin lesions were resolved. Blood tests revealed that the platelet count increased to 99×103/µL two weeks after her discharge and returned to the normal range a month later. The patient is being followed up regularly at our outpatient rheumatologic clinic and shows no recurrence of skin lesions or any other organ involvement for three years. In a recent blood test for aPL, LA was positive, and anticardiolipin and anti-β2-GPI were negative.
DISCUSSION
We reported the case of a 14-year-old girl with catastrophic APS. She was diagnosed with catastrophic APS with SLE on the basis of skin necrosis, small artery thromboses of lower extremities, lupus enteritis, acute MI, acute myocarditis, and positive aPL. The patient was confirmed as aPL-positive at least twice, 10 weeks apart. She was treated with corticosteroids, IVIGs, plasmapheresis, and anticoagulants. However, there was no improvement, and therefore, rituximab was administered. After rituximab treatment, no further thrombotic events have occurred during last 3 years of follow-up.
Currently, there are no standard recommendations or guidelines for the treatment of catastrophic APS because it is a very rare disease in both adults and children [7]. Anticoagulation therapy is the first-line and the most important step in the treatment of catastrophic APS. Antiplatelet therapy can be used in addition to anticoagulant therapy, particularly in patients with arterial thrombotic events [8]. According to the 8th American College of Chest Physicians (ACCP) guidelines, therapeutic and prophylactic uses of LMWH are based on the anti-factor Xa assay (anti-FXa) ranges of 0.5~1.0 U/mL and 0.1~0.3 u/mL, respectively. The ACCP guidelines suggest prophylactic doses of 0.75 mg/kg and 0.5 mg/kg every 12 hours for neonates <2 months and children older than 2 months, respectively, to bring anti-FXa levels in the therapeutic range. In addition, the guidelines for treatment of thrombosis with enoxaparin are based on a study which recommended a dose of 1.5 mg/kg every 12 hours for neonates <2 months and 1.0 mg/kg every 12 hours for children older than 2 months [9]. LMWH at a dose of 1.0 mg/kg was administered every 12 hours to our patient in the present study. There is a linear relationship between the anti-FXa activity and aPTT with a good correlation, and thus, aPTT may be suitable for monitoring thrombosis prophylaxis in patients at high risk of accumulation of LMWH [10]. We are accordingly adjusting the dose of LMWH through the aPTT level. Second-line treatments include plasmapheresis and/or immunomodulatory therapies. In the present study, we performed seven rounds of plasmapheresis during the third admission. Despite these second-line therapy measures, no clinical or laboratory improvements were seen in our patient. This indicated the possibility of refractory catastrophic APS. Rituximab has recently been proven effective in treating catastrophic APS in patients unresponsive to conventional treatment [1112]. In our patient in the present study, rituximab was administered because there was no improvement despite first- and second-line treatments. Rituximab administration caused a dramatic effect in the course of the disease, stopping the inflammatory process. Thereafter, all clinical and laboratory findings showed improvements, and remission is being maintained.
Based on our and previous studies, rituximab may be considered as a treatment for SLE [13]. However, further study is needed to elucidate therapeutic effect and safety data of rituximab on catastrophic APS associated with SLE.
SUMMARY
Catastrophic APS is a rare, severe, and life-threatening condition with a high mortality rate. Thus, early recognition and aggressive therapy are very important for successful treatment. This study is the first report of pediatric catastrophic APS treated with rituximab in Korea. Based on the recent data from several case reports considering the use of rituximab in aPL-positive patients, and results from the present review, rituximab has been used successfully in the treatment of aPL-positive patients, particularly those with refractory catastrophic APS.


 XML Download
XML Download