

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Idiopathic granulomatous hypophysitis (IGH) is a chronic inflammatory lesion of the pituitary gland and the second most common type of hypophysitis. IGH may mimic a pituitary adenoma or craniopharyngioma on MRI and usually present with insidious onset of symptoms including headaches, visual impairment, hormonal deficiencies, and cranial nerve palsies. On the other hand, a rapid onset of neurologic and visual symptoms in an IGH patient is rare. We present the case of rapid onset IGH treated successfully with glucocorticoids.
CASE REPORT
This study was approved by the institutional review board of the author's institution and an informed consent was obtained.
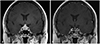
A 41-year-old female presented with a sudden onset of bilateral temporal headaches, blurred vision and diplopia that began four days previously. Neurologic exams revealed left abducens nerve palsy. On further inquiry, the patient recounted that she had mild headaches, occasional galactorrhea and irregular menstruations for the previous two months. An MRI scan of the sella revealed a 2.2×1.7 cm sized gadolinium-enhancing sellar mass with an increase in the mass size and further thickening of the infundibulum compared to the previous MRI performed one month prior (Fig. 1A).
Laboratory evaluations showed an elevated serum prolactin level, measuring 2,217.4 pmol/L (normal range 165–1,010 pmol/L). Basal hormone studies revealed a low cortisol level of 44.14 nmol/L (140–690 nmol/L), a low luteinizing hormone (LH) at 0.8 IU/L (1–104 IU/L), a normal follicle-stimulating hormone (FSH) level of 3.1 IU/L (1–100 IU/L) and a low estradiol level of 18.36 pmol/L (110–1,470 pmol/L). Her serum prolactin level was elevated to 1,652.2 pmol/L (165–1,010 pmol/L). Thyroid function tests showed normal T3, T4 and low thyroid-stimulating hormone (TSH) levels at 2.14 nmol/L (0.92–2.76 nmol/L), 10.68 pmol/L (12–30 pmol/L), and less than 0.05 mIU/L (0.4–5 mIU/L), respectively. Insulin-like growth factor 1 (IGF-1) level was decreased to 14.54 nmol/L (18–60 nmol/L). Rapid adrenocorticotropic hormone (ACTH) test results revealed hypocortisolism, with a basal cortisol level of 35.86 nmol/L, a 30-minute cortisol level of 184.8 nmol/L, and a 60-minute cortisol level of 190.4 nmol/L.
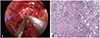
The patient then underwent an endoscopic transsphenoidal biopsy of the hypophysis (Fig. 2A). Permanent biopsy revealed chronic granulomatous inflammation with central caseous necrosis and multinucleated giant cells (Fig. 2B). An acid-fast staining (AFB) stain, mycobacterium tuberculosis nested polymerase chain reaction (PCR) and non-tuberculous mycobacteria-polymerase chain reaction (NTM-PCR) results were negative. IgG4 level was 8.5 mg/dL, which was within normal range. Human chorionic gonadotropin and alpha-fetoprotein levels were assessed for the possibility of germ cell tumors, and were found to be within the normal range. Interestingly, the patient displayed an elevation of angiotensin-converting enzyme level at 1,375.03 nkat/L (normal <670 nkat/L), C-reactive protein level at 10.10 nmol/L (normal 0.76–28.5 nmol/L) and lactate dehydrogenase level at 3.99 µkat/L (normal, 1.7–3.4 µkat/L). Further rheumatologic evaluations for suspected giant cell arteritis due to the elevated ACE level yielded negative results. Serologic workups for syphilis, hepatitis B, hepatitis C or HIV were also negative. Repeat testing of IgG4 level was normal (7.6 mg/dL), and myeloperoxidase-targeting antineutrophil cytoplasmic antibodies and proteinase-3-targeting anti-neutrophile cytoplasmic antibody (ANCA) studies were all negative. The positron emission tomography-computed tomography for other malignancy displayed no hypermetabolic lesions except for the sellar lesion.
The patient recovered without any postoperative neurologic deficits. Postoperative diabetes insipidus was well controlled with intravenous administrations of desmopressin and later on, oral forms of desmopressin. She was given 15 mg of oral prednisolone daily for 3 months, and then tapered to a 15 mg/day of oral hydrocortisone. A postoperative MRI scan performed after three months revealed a markedly decreased residual pituitary gland with mildly thickened infundibulum (Fig. 1B).
DISCUSSION
Granulomatous hypophysitis is a rare pituitary disease, and it is the second most common type of hypophysitis following lymphocytic hypophysitis [1234]. The incidence is estimated to be 1 case per 9 million people per year [5]. The prevalence of granulomatous hypophysitis is currently not precisely reported [46]. Granulomatous hypophysitis has a female preponderance, and is usually diagnosed in the fourth or fifth decade [47].
Granulomatous hypophysitis is histologically differentiated from lymphocytic or xanthomatous hypohysitis by the presence of multinucleated giant cells with caseous necrosis [4]. Granulomatous hypophysitis can also occur as a secondary phenomenon due to other etiologies such as tuberculosis, sarcoidosis, syphilis, fungal infections, pituitary adenomas, Langerhans cell histiocytosis, Erdheim-Chester disease, Wegener's granulomatosis, and giant cell arteritis [2891011]. Therefore, IGH can be diagnosed after exclusion of other secondary causes.
The present case had typical demographic characteristics of IGH. However, our patient complained of sudden severe headache, blurred vision, and diplopia. The most common presenting symptom in IGH is headache followed by menstrual changes, visual changes, and diabetes insipidus, but they are usually of insidious onset [4]. IGH cases with rapid onset symptoms have rarely been reported. Husain et al. [5] described a case of IGH manifesting as clinical pituitary apoplexy, and gross total resection improved the patient's visual symptoms postoperatively. The symptoms may be caused by hemorrhage or infarctions of the pituitary gland, which expands rapidly [4].
Imaging findings of IGH include a symmetric enlargement and homogeneous gadolinium enhancement of the pituitary gland, infundibular thickening, and normal sellar size [241213], and these were observed in our case as well. However, such findings may be seen in other diseases such as pituitary tuberculoma [21213]. Sellar enhancement seen in tuberculoma differs from IGH in that it has central hypointensity with an irregularly enhancing rim, and additional tests such as an interferon gamma release assay test is needed for diagnosis [114]. Apart from aforementioned imaging findings, IGH can also appear as cystic lesions mimicking abscesses [41516]. IgG4-related hypophysitis may also appear similar to IGH. The differential diagnosis should be made by a histological finding of sellar mass or extracranial diseases, high serum IgG4 levels, and clinical improvement with corticosteroid use. Thus, biopsy of the hypophysis is crucial in confirmation of diagnosis.
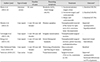
Treatment options of primary hypophysitis include the administration of corticosteroid, surgical excision, and radiosurgery [1718]. The goal of treatment is to alleviate symptoms via hormone replacement and to decompress neurovascular structures [19]. Overall, the symptom resolution was achieved in 80% of IGH patients [4]. In a literature review of 82 cases, Hunn et al. [4] noted that the symptom resolution, hypopituitarism, and recurrence rate was not different between patients who had surgical excision and those who had a biopsy and steroid treatment. However, when patients were treated with surgical excision combined with steroid treatment, the outcome was poorer than when treated with surgical excision only. In addition, there was a case report of IGH unresponsive to prednisolone use [20]. A recent case series by Ved et al. [21] containing 6 patients with pituitary xanthogranuloma reported gross total resection in all patients, and no postoperative recurrence. However, intraoperative pituitary stalk injury led to permanent steroid replacement in 67% of patients, and rendered 4 out of 6 patients thyroxine-deficient [21]. On the other hands, recent case reports culminated in the alleviation of neurologic symptoms after corticosteroid use, and more studies favored corticosteroid treatment over excisional strategy alone (Table 1) [181922]. Corticosteroid replacement in our patient unmasked diabetes insipidus and led to resolution of neurological symptoms.
IGH may be difficult to suspect from the initial presentation. As clinical or imaging diagnostic criteria may be ambiguous and overlap with other etiologies, histologic diagnosis is crucial in confirmation of diagnosis [23]. Establishment of diagnosis is the most important step in order to initiate corticosteroid therapy and prevent delays due to misdiagnosis as pituitary tumors or abscess. Prevention of unnecessary surgical intervention and possible postoperative morbidities may improve outcomes in patients with IGH.
In conclusion, glucocorticoid replacement may result in clinical resolution of IGH without the need for complete surgical resection. A biopsy of the lesion is required for diagnosis of this rare disease entity. Although the presentation of IGH is more common with an insidious onset of symptoms, patients can present with rapid onset of symptoms. Clinical resolution can be achieved by corticosteroid replacement. Thus, a heightened sense of suspicion for IGH is required in a patient presenting with a pituitary mass and rapid onset of neurologic symptoms to prevent unnecessary surgical intervention. Further research is required to elucidate the prevalence, prognostic and risk factors, diagnostic criteria, and optimal treatment options in IGH.
 XML Download
XML Download