

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening hyperinflammatory syndrome related to excessive immune activation of macrophages and T cells [1]. It is characterized by fever, splenomegaly, cytopenia, hypertriglyceridaemia, hypofibrinogenemia, low or absent natural killer (NK) cell activity, elevated blood levels of the soluble IL-2 receptor and ferritin, and haemophagocytosis in biopsy samples of bone marrow (BM), spleen or lymph nodes [12]. Because these clinical and laboratory findings are often present in various diseases, the diagnosis of HLH may be challenging, especially in early stages of disease. Morphological evaluation of BM aspirates in HLH patients usually shows non-specific abnormal findings such as an increased number of histiocytes and lymphocytes with or without hemophagocytosis and a marked left shift in myelopoiesis [2].
A unique flow cytometric (FCM) finding in Epstein-Barr virus (EBV)-positive HLH patients has been reported: clonal expansion of EBV-infected CD8+ T cells with CD5 or CD7 downregulation [23]. CD5 downregulation on activated CD8+ T cells has also been reported in primary (or familial) HLH patients [4]. Thus, the identification of this clonal T cell population with CD5 or CD7 downregulation might provide important information that would aid in the timely diagnosis of EBV-positive HLH. However, limited data regarding FCM findings for CD8+ T cells are available [23]. Furthermore, the clinical relevance of these cell populations remains unclear; thus, further information regarding T cell abnormalities in EBV-HLH is required. Although T cell immunophenotypes in HLH patients have been described, their utility as an adjunctive diagnostic or a prognostic marker for HLH has not been evaluated. For this purpose, we characterized the downregulation patterns of CD5 or CD7 on activated T cells. Furthermore, we evaluated the laboratory utility of FCM analysis in the initial diagnosis of EBV-HLH by T cell immunophenotyping of BM samples showing hemophagocytosis and examined whether an aberrant T cell immunophenotype in HLH can be used as a prognostic marker.
Go to : 
METHODS
Patients
We retrospectively reviewed medical charts of 60 patients with definite hemophagocytosis in the BM who underwent immunophenotyping of T cells using FCM at Samsung Medical Center, Seoul, Korea, from 2014 to 2017. The follow-up period for patients was between January 2014 and September 2019. The median follow-up duration was 29.8 months (95% confidence interval: 23.3–36.3 months). All patients were evaluated for HLH using the 2004 diagnostic criteria [5]: fever, splenomegaly, cytopenias (affecting at least two of three lineages in the peripheral blood; Hb <90 g/L, platelets <100×109/L, neutrophils <1.0×109/L), hypertriglyceridemia (≥3.0 mmol/L) and/or hypofibrinogenemia (≤1.5 g/L), low or absent NK cell activity, ferritin (≥500 ug/L), soluble IL-2 receptor (≥2,400 U/mL), and a molecular analysis (PRF1 and UNC13D genes). Forty-five patients satisfied five or more of these criteria and were finally diagnosed as having HLH [5]. The remaining 15 were excluded. For an EBV-positive HLH diagnosis, the diagnostic criteria for HLH had to be fulfilled, and EBV infection was determined by either the presence of EBV DNA by viremia (>2,000 copies/mL in serum) or by a positive result for Epstein-Barr encoding region in situ hybridization (EBER-ISH) on a BM section [6].
Patients were categorized into three subgroups. The aforementioned HLH patients with EBV-associated disorders, including EBV infection and EBV-positive lymphoproliferative disorders, were designated as EBV-positive HLH (N=27). HLH patients with other disorders, unrelated to EBV infection, such as autoimmune diseases, infections, and idiopathic cases, were designated as EBV-negative secondary HLH (N=15). More detailed characteristics and laboratory findings of each subgroup are shown in Table 1. HLH patients with genetic mutations were designated as familial HLH (N=3) by detecting pathogenic variants in the UNC13D gene (Table 2). This study was approved by the Institutional Review Board of Samsung Medical Center, Seoul, Korea (SMC 2017-12-095), which waived the need for informed consent.
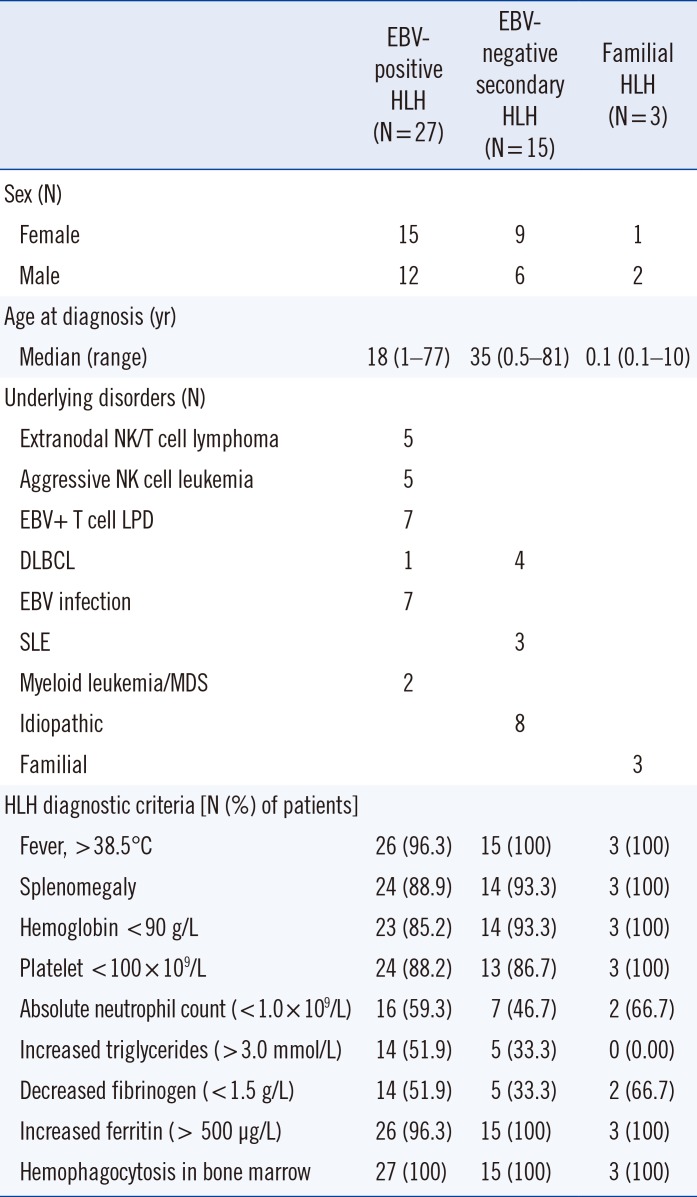

Table 1
Demographics of HLH patients
![]()
Table 2
UNC13D mutations in three Korean patients diagnosed as having familial hemophagocytic lymphohistiocytosis
| Age at diagnosis | Sex | Mutant allele 1 | Mutant allele 2 | Outcome | Reference |
|---|---|---|---|---|---|
| 10 years | Female | c.544C > T (p.Pro182Ser) | c.754-1G > C | Survived | This study |
| 14 days | Male | c.118-308C > T | c.754-1G > C | Survived | Shin et al.[3] |
| 35 days | Male | c.754-1G > C | c.754-1G > C | Died | This study |
![]()
FCM
FCM was performed immediately on fresh BM aspirates in all patients. The samples were collected in EDTA or heparin anticoagulant and routinely processed using a red cell lysis method. Cell suspensions were incubated with combinations of eight monoclonal antibodies (Becton Dickinson, San Jose, CA, USA): CD45-AmCyan, CD3-allophycocyanin-cyanine 7, CD4-pacific blue, CD8-PeCy7, CD56-allophycocyanin, CD2-peridinin chlorophyll protein, CD5-fluorescein isothiocyanate, and CD7-phycoerythrin. Data were acquired using a FACSCanto II flow cytometer (Becton Dickinson) and analyzed using the Kaluza software (Becton Dickinson). CD5 or CD7 downregulation on T cells was determined by adjusting the cutoff range of the non-neoplastic T cell populations reported by Jamal et al. [7]. Three reviewers confirmed the agreement of all analyzed FCM data.
Statistical analysis
Categorical variables were expressed as frequencies and percentages, and continuous variables were expressed as medians with ranges. The cut-off values of the categorical variables were taken from the 2004 diagnostic criteria for HLH [5].
Survival curves were generated using the Kaplan-Meier method, and survival rates were compared using the log-rank test. Overall survival was measured from the date of diagnosis to the date of all-cause death and was censored at the date of the last follow-up visit. All analyses were performed using SPSS for Windows, version 23 (SPSS Inc., Chicago, IL, USA). P≤0.05 (two-tailed) was considered statistically significant.
Go to :
RESULTS
FCM
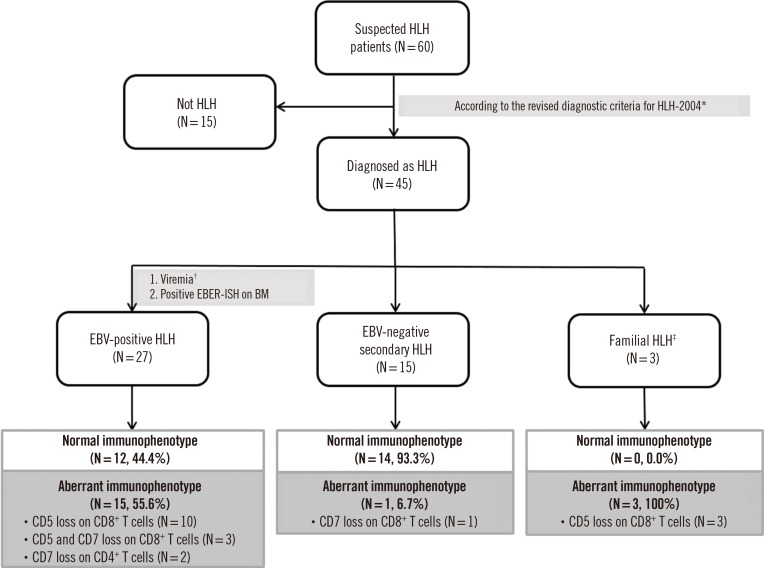
The aberrant immunophenotype patterns observed in the three subgroups are shown in Figs. 1 and 2, and Table 3. Aberrant immunophenotypes, including CD5 and/or CD7 downregulation on CD4+ helper T cells or CD8+ cytotoxic T cells, were observed in 55.6% (15/27) of EBV-positive HLH patients, 6.7% (1/15) of EBV-negative secondary HLH patients, and 100% of familial HLH patients (3/3).
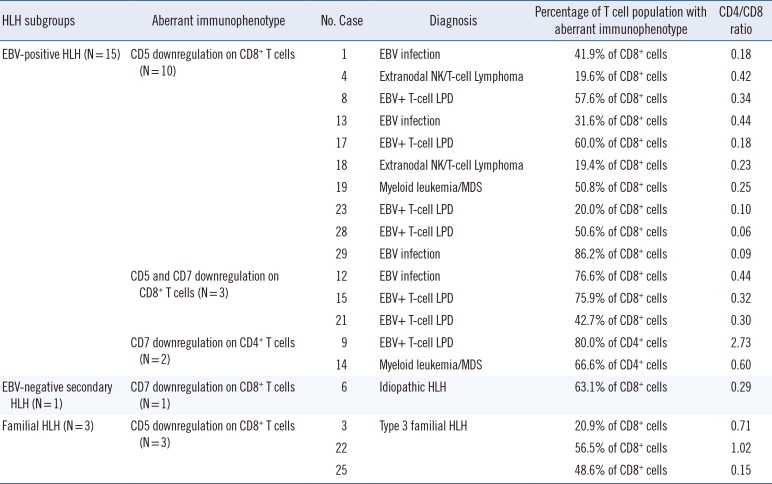
 | Fig. 1Study design flowchart for classification of HLH patients.
*HLH patients were identified according to the 2004 criteria [13]; †>2,000 genome copies/mL of serum; ‡Three familial HLH cases were diagnosed as type 3 familial HLH based on detection of pathogenic variants of the UNC13D gene.
Abbreviations: HLH, hemophagocytic lymphohistiocytosis; EBV, Epstein-Barr virus; EBER-ISH, Epstein-Barr encoding region in situ hybridization; BM, bone marrow; MDS, myelodysplastic syndromes; LPD, lymphoproliferative disease.
|
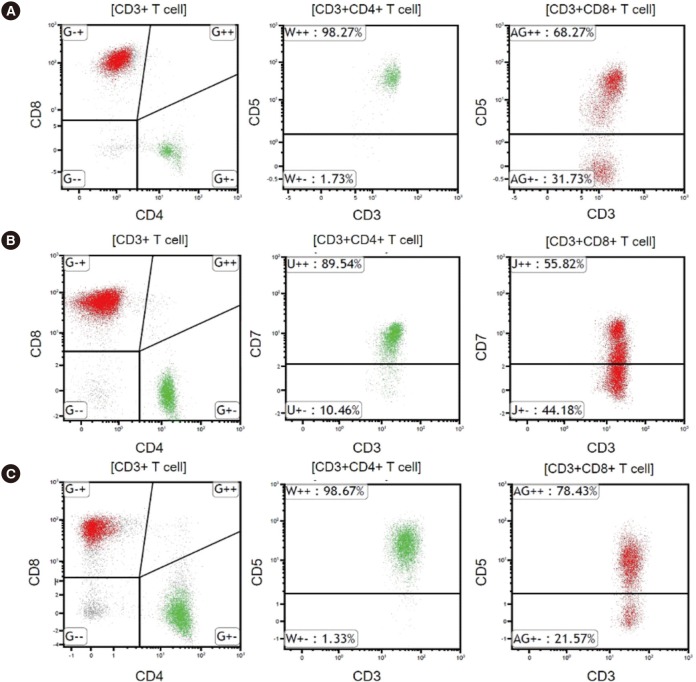 | Fig. 2Representative flow cytometric findings in HLH patients. Red and green colors indicate CD8+ T cells and CD4+ T cells, respectively.(A) CD5 downregulation on CD8+ T cells in EBV-positive HLH. (B) CD7 downregulation on CD8+ T cells in EBV-negative secondary HLH. (C) CD5 downregulation on CD8+ T cells in familial HLH.
Abbreviations: HLH, hemophagocytic lymphohistiocytosis; EBV, Epstein-Barr virus.
|
Table 3
Flow cytometric findings for HLH subgroups with an aberrant T cell immunophenotype
![]()
Overall survival
Overall survival did not differ significantly between patients with normal and abnormal immunophenotypes. Subgroup analysis showed that aberrant loss of CD5 or CD7 was not significantly related to overall survival in EBV-positive HLH and EBV-negative secondary HLH patients (Fig. 3).
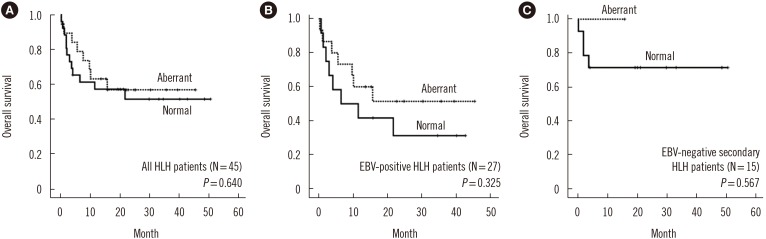 | Fig. 3Kaplan-Meier survival analysis of HLH patients based on the presence of an aberrant T cell immunophenotype. (A) Overall survival of all HLH patients. (B) Overall survival of EBV-positive HLH patients. (C) Overall survival of EBV-negative secondary HLH patients.Abbreviations: HLH, hemophagocytic lymphohistiocytosis; EBV, Epstein-Barr virus.
|
Go to :
DISCUSSION
HLH can be categorized as primary or secondary based on the underlying cause. Primary HLH is associated with a proven genetic etiology or a family history and accounts for the majority of pediatric HLH cases [89]. Familial HLH is caused by mutations in several genes, including PRF1, UNC13D, STX11, and STXBP2. Approximately 70% of familial HLH cases belong to type 2 and type 3 familial HLH, which are caused by PRF1 and UNC13D, respectively [1011]. UNC13D is the predominant causative gene in Korean familial HLH patients [31213]. In our study, all three familial HLH patients were diagnosed as having type 3 familial HLH with a known splicing mutation c.754-1G>C. HLH also occurs secondary to various underlying disorders, including malignancies, rheumatologic diseases, and various infections [1415]. Among infections, EBV is the most common virus triggering the occurrence of HLH [161718]. Therefore, we categorized 45 HLH patients into familial HLH (N=3) and secondary HLH, which was reclassified into EBV-positive HLH (N=27) and EBV-negative HLH (N=15), according to EBV infection status.
EBV infection is usually asymptomatic but occasionally leads to a variety of diseases, such as acute infectious mononucleosis, chronic active EBV infection, EBV-infection-associated HLH, EBV-associated B cell lymphoproliferative disorder (LPD), and EBV-associated T/NK cell LPD [1920]. In the HLH response to EBV, T cells are the major target of EBV infection, leading to polyclonal or monoclonal proliferation of EBV-infected T cells [721]. In infectious mononucleosis, a reactive CD8+ T cell population with dim or absent CD7 is commonly found [22]. Wada et al. [23] reported that a lack of CD5 expression on CD8+ T cells constitutes a unique immunophenotypic feature of EBV-positive HLH. We also identified 15 patients with downregulation of CD5 and/or CD7 expression on T cells in the EBV-positive HLH group, similar to previous reports [2123], and an aberrant T cell immunophenotype was less frequent in EBV-negative HLH than in EBV-positive HLH; 93.3% (14/15) of the EBV-negative secondary HLH patients showed normal T cell immunophenotypes. Our findings extend previous findings that EBV infection is associated with downregulation of CD5 and/or CD7 expression on T cells.
Aberrant T cell immunophenotypes have been reported mostly in patients with EBV-HLH but also in patients with familial HLH [34]. Karandikar et al. [24] revealed expanded subpopulations of CD8+ T cells with the lack of CD5 expression in a patient with familial HLH and showed no evidence of bacterial or viral infection, including cytomegalovirus (CMV) and EBV. Shin et al. [3] also reported a Korean patient with familial HLH with an abnormal population of CD8+ T cells with downregulated levels of CD5 or CD7 and T cell monoclonality and no laboratory evidence of active EBV or CMV infection. Interestingly, aberrant T cell immunophenotypes have been reported in familial HLH patients without evidence of active EBV infection. Wada et al. [4] also found that the activated CD8+ T cells exhibited down-regulation of CD5 and that CD4+ T cells from the patients with FHL2 exhibited normal expression of CD5, although they did not provide any laboratory evidence of active EBV infection. This unique subpopulation is thought to be the result of dysregulated proliferation of CD8+ T cells, similar to clonal proliferation of EBV-infected cells [25]. Consistent with previous reports, we found a lack of CD5 on CD8+ T cells in all three familial HLH patients (as well as one case from reference [3]) without evidence of active EBV infection [426]. These data suggest that the lack of CD5 on CD8+ T cells in familial HLH occurs in various situations other than EBV infection.
Some T cell immunophenotypes can be used to identify clonal T cell disorders, such as skewing of the CD4:CD8 ratio, loss of surface CD4 or CD8, co-expression of CD4 and CD8, and loss of CD5 and/or CD7 [27]. Among them, a lack of CD5 expression on CD8+ T cells could serve as a useful marker of T cell proliferation in EBV-positive HLH. However, EBV infects T cells in EBV-HLH, leading to polyclonal or monoclonal proliferation of EBV-containing T cells, which may mimic T cell LPD (T-LPD) [21]. Thus, careful interpretation is required for the immunophenotyping and clonality data in T cell proliferation in association with EBV-HLH, to avoid erroneous diagnosis of T-LPD. In our study, the aberrant T cell immunophenotype in HLH was helpful in discriminating EBV-negative secondary HLH and EBV-positive HLH, but it may not be useful for discriminating EBV infection in HLH from T-LPDs (data not shown).
The prognosis of EBV-positive HLH may be better than that of familial HLH [282930]. Imashuku et al. [17] performed a longitudinal follow-up study on EBV-positive HLH patients and showed that the majority of effectively treated EBV-positive HLH patients had a good prognosis, except for a few cases of early death. We evaluated whether aberrant T cell immunophenotypes in HLH can be used as a prognostic marker. We did not find a significant association between survival and aberrant loss of CD5 or CD7 expression on T cells in both EBV-positive HLH and EBV-negative secondary HLH patients. However, aberrant loss of CD5 or CD7 expression on T cells tended to confer a survival benefit among EBV-positive HLH patients, although this tendency was not statistically significant. This pattern was repeated among EBV-negative secondary HLH patients. Our findings imply that immunophenotyping in patients with secondary HLH might help proactively discriminate patients showing better outcomes. However, more data are needed to determine the prognostic significance of aberrant T cell immunophenotypes in HLH patients.
In conclusion, our study suggests that the aberrant T cell immunophenotypes in HLH may assist in discriminating EBV-negative secondary HLH and EBV-positive HLH, but they may not be useful as a prognostic marker. Despite the limitation of a relatively small sample size, our study extends previous findings that the aberrant T cell immunophenotypes are more frequent in EBV-positive HLH than in EBV-negative secondary HLH.
Go to :
 XML Download
XML Download