

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Creutzfeldt-Jakob disease (CJD) is the most frequent human prion disease, although it is rare. The vast majority of CJD cases are sporadic (about 85%) and calculated prevalence of it is approximately 1-1.5 cases per one million population a year with a worldwide distribution [1]. In Korea, the social anxiety increased a lot regarding import of beef from U.S. in 2008. As a result, CJD including variant CJD has become the center of public interest in Korea. It requires professional experience in the course of diagnosis, and neurologist plays a critical role in the disease surveillance as well as in the diagnosis and management of it. The registration rate and accuracy of diagnosis in Korea are still very low compared to that of developed countries. This study was aimed to investigate the level of awareness, clinical experience and preparedness of Korean neurologists who would encounter the patient of CJD at first place. This study would be used as basic data for improving Korean surveillance system and also for better caring and educational system.
METHODS
The survey was performed for the participants on the 31st Annual Meeting of the Koran Neurological Association in April 2012. Total participants of the meeting were 688 (442 specialists, 246 residents) and 133 neurologists voluntarily responded survey among them. A survey was considered to be the most efficient and accurate measure to evaluate neurologists' current knowledge and practice regarding CJD. The survey questions were developed with 29-item questionnaire (appendix) covered 5 categories which were demographics of neurologists, clinical experience on CJD, diagnosis, terminology and educational/reporting system. It was self-administrated survey and after reviewing of collected data, descriptive statistics, such as percentages and sample size were used to describe how neurologists responded to specific questions. Most of the data are analyzed according to specialty of neurology (resident or specialist of neurology).
RESULTS
Demographics of the participating neurologists on this survey
Among the neurologists who participated on the 31st Annual Meeting of the Koran Neurological Association in April 2012, 133 members replied on our survey. Eighty-three members were specialists of neurology and 51 members were residents of neurology. Age range of participants distributed from 20s to 50s. About 75% of respondents worked for university hospital. The most frequent city they worked in was Seoul (53.4%) and Gyeonggi-do held the second rank (17.3%) (Table 1).
Clinical experience on CJD
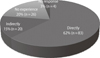
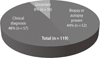
On this survey, 62.4% of the neurologists had cared CJD suspected patients directly and 15% had observed diagnosed cases in their hospital although they had not cared in person, and 19.5% had not cared CJD suspected patients before (Fig. 1). The percentage of the neurologists who made clinical diagnosis of CJD were 48% (n=57) and who made confirmatory diagnosis by brain biopsy or autopsy were 44% (n=52) (Fig. 2). Among respondents, 27.1% of them said that CJD might occur more than 40 cases a year and 24.8% said that 10 to 19 cases might occur in Korea. Among 27.8% of neurologists answered that clinically diagnosed CJD might be less than 10 cases per year and 24.8% replied 10 to 19 cases a year and just 16.5% considered more than 40 cases a year in Korea (Table 2).
Diagnosis
Whereas 48% of neurologists were confident in the diagnosis of CJD clinically unless it was atypical case, 44% were not confident. The Lack of clinical experience and limited information were the most difficult problems regarding the diagnosis of CJD (51.9%) and various clinical presentations in the early stage of disease were also playing a role as an obstacle for correct diagnosis (45.1%). Respondents thought that most important diagnostic clues for suspecting CJD were rapid progressive cognitive decline (86.5%) and myoclonus (10.5%). Regarding useful diagnostic tools in order of importance were suggested that clinical findings (66.2%), brain MRI (45.1%), CSF 14-3-3 protein (30.1%), EEG (36.8%) and gene (PRNP) test (42.9%) in order. When positive finding of CSF 14-3-3 protein is combined with rapidly progressive dementia, 31.6% of respondents answered that the possibility of CJD is about 60 to 79% and 28.6% answered as 40 to 59%. If myoclonus is added to them, 40.8% of neurologists regarded the possibility of CJD is increased as high as 80 to 100% and 28.6% regarded as 60 to 79%. They responded that CSF 14-3-3 protein could be negative in 63.2% and 22.6% of them thought that initially it could be negative but it would be converted into positive in the course of disease. About 31% of neurologists estimated positive predictive values of CSF 14-3-3 protein in Korea might be 40 to 59%, 19.5% of them as 60 to 79% and 17.3% of them as 20 to 39% respectively. With regard to the question about the specificity of diagnostic tools, positive findings of clinical signs, specific brain MRI findings, CSF 14-3-3 protein and EEG periodic sharp wave complex were thought to be found in other diseases also (89.5%, 73.7%, 83.5%, 91.7% respectively). About 50% of neurologists answered that brain biopsy or autopsy was necessary just in case of confusion in the diagnosis of CJD whereas 45.1% replied that it was necessary although it was certain as CJD clinically. Similarly to this, 52.6% of respondents said they would transfer just in case of confusion in the diagnosis of CJD for brain biopsy and 46.6% answered that they would definitely persuade caregiver for brain biopsy. Regarding transferring CJD suspected patients to superior hospitals for definite diagnosis or second opinion, 35.3% said they had transferred most of the patients for second opinion, 32.3% said that they hadn't transferred unless caregiver wanted to and 30.8% replied just in case of confusion in the diagnosis (Table 3).
Terminology and awareness
About the terminologies related to CJD, most of the respondents answered that they know well or know a little at least about sporadic CJD (sCJD), variant CJD (vCJD), iatrogenic CJD (iCJD) and familiar CJD (fCJD) (49.6%, 41.4%, 43.6%, 39.8% respectively). Regarding diagnostic criteria of sCJD by National CJD surveillance, Heidenheim variant CJD, 6 molecular subtypes of sCJD, PSPr (proteinase sensitive prionopathy)/VPSP (variably PSPr), and Heidenheim variant CJD, small proportion of them answered that they know well or know a little at least about those disease entities or criteria (37.6%, 41.4%, 45.9%, 21.8% respectively) (Fig. 3). Many respondents replied that sCJD, iCJD and fCJD actually had occurred in Korea before (94.7%, 75.2%, 44.4% respectively). However, only 49.6% thought that vCJD had not occurred and most of neurologists said they didn't know whether PSPr, GSS, FFI, or sFI occurred or not in Korea (50.4%, 60.2%, 50.4%, 63.2% respectively) (Fig. 4).
About 8.5% of neurologists haven't used terms "human mad cow disease" to explain sCJD but 18.8% had used it. The 60.9% of respondents said that term should not be used but 36.8% replied that it was not proper but it could be used to explain sCJD. Most of the neurologists (66.2%) had been asked about medical issues related to CJD and they are general public (33.1%), paramedics (21%) and doctors of other specialty (20%) (Table 4).
Educational/reporting system
About 44% of neurologists on this survey had heard of the reporting system of suspected CJD patient to public health center but don't know well about it, 34.1% are well aware of it and 11.3% said that they never heard of it. Most of the respondents had participated in the lecture or educational programs related to CJD 1 to 2 times (52.6%) and 24.1% participated more than 3 times but 21.8% never had chance to participated in them. About the question asking the necessity of increasing educational opportunity for neurologists, 67.7% of respondents said "yes" but 30.8% of them thought it wss still enough. About 72.9% of respondent replied that they would participate in the educational program as far as circumstances permit and 23.3% said they would participate for sure. Regarding partial economical supporting system for medical expenses of CJD patients as rare and incurable disease, 45.1% of respondents said that they had never heard of that and 37.6% had heard of it but don't know well and the proportion of the respondents who knew about it was as low as 16.5% (Table 5).
DISCUSSION
According to the reported incidence of CJD, it seems to be 50 to 75 patients per year in Korea. Although the incidence is very low, it is important as a fatal disease without cure and should be considered as first differential diagnosis of patients with rapidly progressive dementia. In 2011, the first Korean case of iatrogenic CJD was reported which also increased the public interest among the people [2]. So, the possibility of transfection to other persons by medical devices, operational tools or transfusion also should be considered.
On this survey about 44% of neurologists (n=52) encountered CJD patients confirmed by biopsy or autopsy (Fig. 2) and it is very high percentage when we consider the low rate of biopsy or autopsy performance in Korea. The reason why it showed high proportion might be the overlapping patients with CJD among the respondents. Twenty-seven percent of respondents answered that CJD might occur more than 40 cases a year but 52.6% of them considered clinically diagnosed CJD would be less than 20 cases, so most neurologists recognized the number of real occurrence of CJD is larger than that of clinically diagnosed cases in Korea. Actually, according to the reports of KCDC, about 30 cases of sporadic CJD have been reported since 2008 annually but estimated occurrence rate of CJD is more than 50 to 60 cases a year in Korea [3]. We do not know the real number of CJD cases because the definite diagnosis of CJD is very rare, and possible diagnosis seems not so valid in Korea. Large proportion of Korean neurologists regarded the incidence as lower than that of calculated incidence. About 62% of the neurologists on this survey had experience of caring CJD patients personally. When we added the number of respondents who just observed CJD patient diagnosed in their hospital although they did not care personally, 77.4% of neurologists had experienced CJD patient directly or indirectly. So, most of the Korean neurologists who participated in the survey had experience of CJD although it is very rare disease.
Rapidly progressive dementia and myoclonus would be the main clinical signs of sporadic CJD [4]. On this survey, 86.5% of respondents answered that rapidly progressive dementia would be the most important clinical sign and 10.5% of respondents replied myoclonus would be. Question about the order of clinical usefulness as diagnostic tool was as follows - clinical signs, CSF 14-3-3 protein, brain MRI, gene test and EEG in order. Among them, brain MRI was not included in revised 1998 diagnostic criteria of World Health organization for sCJD but it is currently modified to incorporate MRI feature [5, 6]. Brain MRI showed higher sensitivity and specificity (96% and 93% respectively) in diagnosis of CJD [7] over CSF 14-3-3 protein (sensitivity 86%, specificity 68%)[8] or EEG (sensitivity 64%, specificity 91%)[9]. Regarding question asking diagnostic specificity of each tool on the basis of 95% specificity, most of the neurologists answered that any positive finding of them would be found in other diseases too. So lack of definite diagnostic tool could be one of the causes of increasing difficulty in the diagnosis of CJD in clinical setting. CSF 14-3-3 protein shows low specificity because it can reveal false positive results in other disease such as herpes simplex encephalitis, hypoxic brain damage, brain metastasis, paraneoplastic syndrome or metabolic syndrome [10]. So other CSF markers such as tau protein or S100b protein are used combined with CSF 14-3-3 protein in some laboratories of other countries [11]. By using cell culture of neurons and glial cells, it is suggested that CSF 14-3-3 protein is just a marker of injured brain tissue rather than that of CJD [12]. The annual number of suspected CJD patients referred to KCDC is increasing constantly from 51 cases in 2008 to 91 cases in 2010. The positive ratio of CSF 14-3-3 protein among referred cases is more than 50% [3]. About 50% of them who showed CSF 14-3-3 positivity are reported as CJD in Korea. Final diagnosis of other cases who showed CSF 14-3-3 protein positive are composed of infectious disease (43%), toxicmetabolic disease (23%), epileptic disorder (10%), tumor (7%) and others (15%) (in submission) . This was in accordance with the result of answers from neurologists who estimated positive predictive value of CSF 14-3-3 protein was from 40 to 59%. When positive finding of CSF 14-3-3 protein is combined with rapidly progressive dementia, 31.6% of respondents answered that the possibility of CJD would be about 60 to 79%. If myoclonus is added to them 40.8% of neurologists regarded the possibility of CJD would increase as high as 80 to 100%. So, rapid progressive dementia, myoclonus and CSF 14-3-3 protein play major role in the diagnosis of CJD in Korean clinical setting. Many neurologists (44%) were not confident in the diagnosis of CJD and it was more evident in residents (62.7%) than specialists (31.7%). Two main problems of this diffidence might be due to the lack of clinical experiences because CJD is rare disease (51.9%) and it showed non-specific initial clinical findings (45.1%). So it suggested that there is in need of educational programs to relieve these problems especially for residents. Many neurologists (35%) would transfer CJD suspected patient to other superior hospital for further evaluation or second opinion. This result might be derived from the burden of confirmative diagnosis as CJD which is fatal disease without cure and from difficulties in differential diagnosis because of its variable initial clinical presentations. The most common first sign was reported as cognitive decline such as deficit in attention, memory or judgment [13]. In addition to it, emotional and behavioral change, sleep disorder are also common. Myoclonus, which is especially induced by startling stimulus, might be observed 90% of cases in the course of disease progression. Extrapyramidal symptoms such as hypokinesia or cerebellar symptoms such as nystagmus, ataxia could be found in two thirds of the patients and 20 to 40% of cases showed them as main symptoms [13]. For example, it was reported that iatrogenic CJD has a tendency of presenting cerebellar symptoms in the early stage of disease [14]. Pyramidal signs such as increased deep tendon reflex or positive Babinski sign can be found in 40-80% of patients. After following up 52 cases of young sCJD patients aged less than 50 years old, it was reported that psychiatric symptoms could be easily found as it is observed in variant CJD [15]. And there are some subtypes or variant form of CJD regarding main focal neurologic signs. Heidenhain variant CJD which present visual symptoms at first and Oppenheimer-Brownell variant CJD which shows mainly cerebellar symptoms could be examples of them.
About 85 to 90% of neurologists on this survey replied that they know well or know a little at least about sCJD, iCJD, fCJD, and vCJD. On the other hand, regarding Heidenheim variant CJD, PSPr (Proteinase sensitive prionopathy)/VPSP (Variably PSPr), Gerstamn-Strausler-Scheinker syndrome (GSS), fatal familiar insomnia (FFI), sporadic fatal insomnia and 6 molecular subtypes of sCJD, large proportion of them answered that they did not know about those disease entity (76.7%, 53.4%, 60.2%, 50.4%, 63.2%, 58.6% respectively). So, it revealed the necessity of education afterward regarding those rare form of prion diseases. Regarding 6 molecular subtypes of sCJD, one of the possible reasons that it showed low recognition rate among Korean neurologists might be due to low incidence of brain biopsy or autopsy performance because molecular subtype is decided by those studies. In Korea, case of vCJD has never been reported but it was in the center of great social issues across the nation related to import of beef from U.S. in 2008. However, 40 neurologists (30%) believed that vCJD had been reported in Korea and 27 neurologists (20.3%) answered that they didn't know whether it was reported or not. About half of them are specialist working in university hospital. Eleven neurologists answered that there had not been sCJD in Korea or they did not know whether it occurred or not, although 9 of them are residents of university hospital in Seoul. Some of respondents (18.8%) have used the term "human mad cow disease" to explain sCJD and 36.8% of neurologists answered that this term could be usable although they know it is not proper. These results show possibilities of evoking social problems. In Korea, "human mad cow disease" means vCJD but it is not appropriate term and should be modified to other term "CJD related to mad cow disease". The proportions of neurologists who answered that they knew the diagnostic criteria of sporadic CJD suggested by WHO or National CJD surveillance Unit was very low as 19% and 9% respectively which showed low awareness of CJD diagnostic criteria.
For definite diagnosis as CJD, it is required to confirm proteinase K resistant protein in Western blotting and to identify abnormal prion protein in immunohistochemistry on brain tissue. Triad of pathologic finding of CJD are neuronal cell loss, gliosis and vacuolization in the brain and spinal cord [16]. In this survey, 51.9% of neurologists answered that they would recommend brain biopsy for definite diagnosis just in case of confusion in the clinical diagnosis of CJD. On the contrary to it, 45.1% of respondent said that brain biopsy is necessary even though it is clinically certain. The brain tissue is not easily obtainable in Korea so the total number of referred brain tissue for CJD is just less than 5 (2cases in 2011, 5cases in 2010, 4cases in 2009, 2008, 2007) annually so, the number of actual brain biopsy is too small compared with the need of neurological specialists. Contradictory realization about brain biopsy or autopsy in Korea may contribute to the reluctant attitude to it. On top of it, surgeons also show reluctance to perform brain biopsy because of the burden of invasive procedure to this fatal transmissible disease.
Although definite diagnosis of CJD requires examination on brain tissue according to diagnostic criteria, the burden of invasive procedure described above and the possibility of false negative finding when brain tissue with no invasion is acquired also must be considered. University of California, San Francisco (UCSF) proposed diagnostic criteria of CJD based on brain MRI in 2010 [7]. Owing to the usefulness of diffusion-weighed image (DWI), they didn't recommend brain biopsy anymore in most cases of CJD patients and it is performed when it showed uncertain feature after DWI. In Korea, comparative analysis is required to evaluate which combination of diagnostic tools can be the most accurate and efficient method for diagnosis of CJD.
In summary, most of the Korean neurologists have encountered CJD although it is very rare disease. They are well recognized about the annual occurrence of CJD, sensitivity and specificity of diagnostic tools which was nearly the same with that of reported in the literature. They had difficulty in the diagnosis of CJD because of the variable initial clinical presentations in the early stage and the lack of clinical experience especially in residents. They also have some misunderstanding not only in the subtypes of CJD or diagnostic criteria but also in the vCJD which was in the center of great public interest in Korea. These suggested the necessity of educational system for neurologists. The term "Human mad cow disease" should be reconsidered because of its potential possibilities that might evoke social problems. Although neurologists do not seem to be firmly prepared to CJD, 66.2% of them are asked about the CJD from many paramedics, doctors of other specialty or general public. Therefore considering the role of primary information providers of CJD, the educational opportunity for the neurologists seems very important to avoid the delivery of false information. About 67% of respondents also replied that they need more educational opportunity than now. Korean neurological association tried to raise the concern about CJD for neurologists through 2010 CJD symposium and it contributed somewhat to increased number of CJD suspected patients who were referred to Korea Centers For Disease Control and Prevention (KCDC) afterward [5]. This might be the good example which enhanced the concerns about CJD among the neurologists by educational symposium. The essential knowledge about CJD in clinical setting is not so much that regular small workshop for neurological resident or specialist would be adequate. Most of the neurologists are not aware of the reporting system of patients or supporting system for medical expenses, so the regular public relations also seem necessary for neurologists.








 XML Download
XML Download