

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Abstract
Creutzfeldt-Jakob disease (CJD) is a degenerative neurological disorder that is incurable and invariably fatal. It is characterized by rapidly progressive dementia presenting with memory loss, personality changes and hallucinations. The symptoms of CJD are caused by progressive death of neurons in the central nervous system, which is associated with build-up of the abnormal prion proteins forming amyloids. In human, CJD can be acquired genetically through a mutation of the gene encoding for the prion protein (PRNP). This occurs in only 5-10% of all CJD cases. We report a 64-year old woman with CJD carrying a V180I mutation that features late onset, rapid progression, no periodic sharp wave complexes on electroencephalography, and cortical signal change and edema in bilateral frontotemporoparietal lobes and basal ganglia on MRI.
Figures and Tables
Fig. 1
Diffusion weighted MR images show cortical signal change and edema in both frontotemporoparietal lobes and both basal ganglia. Both cerebellum and occipital lobes are spared.
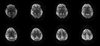
Fig. 2
T2 weighted MRI also shows cortical signal change and edema in both frontotemporoparietal lobes and both basal ganglia. Both cerebellum and occipital lobes are spared.
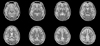
Fig. 3
Brain PET-CT shows marked hypometabolism in bilateral cerebral hemispheres with sparing of the occipital regions and primary motor sensory cortices.

Fig. 4
MR spectroscopy shows decreased NAA peak and increased choline peak in both frontotemporoparietal lobes and both basal ganglia.
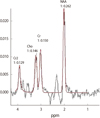
Fig. 6
(A) DNA sequence at codon 180 of the PRNP gene from the patient (left) and a normal control (right). There is a point mutation causing a substitution of GTC(Val) by ATC(Ile) at codon 180. (B) Homozygosity for methionine at codon 129 and homozygosity for glutamate at codon 219 are also identified in the sequence analysis of the PRNP gene mutation of the patient.

References
1. Rabinovici GD, Wang PN, Levin J, Cook L, Pravdin M, Davis J, et al. First symptom in sporadic Creutzfeldt-Jakob disease. Neurology. 2006. 66:286–287.

2. Heinemann U, Krasnianski A, Meissner B, Grasbon-Frodl EM, Kretzschmar HA, Zerr I. Novel PRNP mutation in a patient with a slow progressive dementia syndrome. Med Sci Monit. 2008. 14:CS41–CS43.
3. Kong QK, Surewicz WK, Petersen RB, Zhou W, Chen SG, Gambetti P, et al. Prusiner SB, editor. Inherited Prion Diseases. Prion Biology and Disease. 2004. 2nd ed. Cold Spring Harbor: Cold Spring Harbor Laboratory Press;673–776.
4. Zerr I, Poser S. Clinical diagnosis and differential diagnosis of CJD and vCJD. With special emphasis on laboratory tests. APMIS. 2002. 110:88–98.
5. World Health Organization. WHO Global surveillance, diagnosis and therapy of human transmissible spongiform encephalopathies: Report of a WHO consultation; Emerging and other communicable diseases, surveillance and control. 1998. 02. 9–11.
6. Shiga Y, Miyazawa K, Sato S, Fukushima R, Shibuya S, Sato Y, et al. Diffusion-weighted MRI abnormalities as an early diagnostic marker for Creutzfeldt-Jakob disease. Neurology. 2004. 63:443–449.
7. Young GS, Geschwind MD, Fischbein NJ, Martindale JL, Henry RG, Liu S, et al. Diffusion-weighted and fluid-attenuated inversion recovery imaging in Creutzfeldt-Jakob disease: high sensitivity and specificity for diagnosis. AJNR Am J Neuroradiol. 2005. 26:1551–1562.
8. Yamada M. Prion diseases in Japan: analysis of 918 patients. Rinsho Shinkeigaku. 2007. 47:805–808.
9. Chasseigneaux S, Haik S, Laffont-Proust I, De Marco O, Lenne M, Brandel JP, et al. V180I mutation of the prion protein gene associated with atypical PrPSc glycosylation. Neurosci Lett. 2006. 408:165–169.
10. Nixon R, Camicioli R, Jamison K, Cervenakova L, Mastrianni JA. The PRNP-V180I mutation is associated with abnormally glycosylated PrP-CJD and Intracellular PrP accumulations. Presented at XIVth International Congress of Neuropathology Scientific Programme. Brain Pathology. 2000. 10:670.
11. Jin K, Shiga Y, Shibuya S, Chida K, Sato Y, Konno H, et al. Clinical features of Creutzfeldt-Jakob disease with V180I mutation. Neurology. 2004. 62:502–505.
12. Yang TI, Jung DS, Ahn BY, Jeong BH, Cho HJ, Kim YS, et al. Familial Creutzfeldt-Jacob Disease with V180I Mutation. J Korean Med Sci. 2010. 25:1097–1100.
 XML Download
XML Download