

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Fungi produce a wide variety of structurally unique and biologically active secondary metabolites.12 Of the fungal natural products, meroterpenoids, which contain fragments from both terpenoid and polyketide precursors, are widely distributed in several species, in particular, Penicillium and Aspergillus spp.3 The remarkable structural diversity and significant bioactivity of these compounds have attracted significant chemical and biomedical interests.4 One particularly interesting group of fungal meroterpenoids is the tetra- or penta-cyclic austin class. Since austin, the first example, was identified from Aspergillus ustus in late 1970s,5 various compounds of this family of highly oxygenated meroterpenoids have been isolated from both Aspergillus and Penicillium spp.6 Recently, studies on these compounds have been focused on their biosynthesis in which not only a full biosynthetic pathway but also key enzymes with fascinating activities have been defined.789 These studies have significantly contributed to the structural diversity of fungal meroterpenoids and the potential of expression systems in the biosynthesis of fungal natural products.
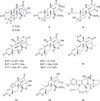
In our search for fungi-derived novel compounds, a strain (strain number FCH061) of Penicillium sp. was isolated from sediment samples collected offshore of Chuja-do, Korea. Based upon the results of LC-ESI-MS analysis of the crude culture extract, the presence of various secondary metabolites prompted us to thoroughly investigate this strain. The large scale cultivation, followed by extraction and separation using a variety of chromatographic methods yielded fourteen compounds (1 – 14) including two new compounds, preaustinoids E (1) and F (2) (Fig. 1). Herein we report the structure determination of these compounds as new austin-type meroterpenoids by a combination of spectroscopic analyses.
Experimental
General experimental procedures
Optical rotations were measured on a JASCO P1020 polarimeter (Jasco, Tokyo, Japan) using a 1cm cell. UV spectra were acquired with a Hitachi U-3010 spectrophotometer (Hitachi High-Technologies, Tokyo, Japan). IR spectra were recorded on a JASCO 4200 FT-IR spectrometer (Jasco, Tokyo, Japan) using a ZnSe cell. NMR spectra were recorded on Bruker Avance 600 spectrometer (Bruker, Massachusetts, USA). 1H and 13C NMR spectra were measured in CDCl3 solutions at 600 and 150 MHz, respectively. High resolution FAB mass spectrometric data were obtained at the Korea Basic Science Institute (Daegu, Korea) and were acquired using a JEOL JMS 700 mass spectrometer (Jeol, Tokyo, Japan) with meta-nitrobenzyl alcohol (NBA) as the matrix. Semi-preparative HPLC separations were performed on a SpectraSYSTEM p2000 equipped with a SpectraSYSTEM RI-150 refractive index detector. All solvents used were spectroscopic grade or distilled from glass prior to use.
Fungal material
The fungal strain Penicillium sp. (strain number FCH061) was isolated from underwater sediment collected off the coast of Chuja-do, Korea, in October, 2012. The sample was diluted using sterile seawater. One milliliter of diluted sample was processed utilizing the spread plate method in YPG medium (5 g of yeast extract, 5 g of peptone, 10 g of glucose, 0.15 g of penicillin G, 0.15 g of streptomycin sulfate, 24.8 g of Instant Ocean, and 16 g of agar in 1 L of distilled water) plates. The plates were incubated at 28℃ for 5 days. The strain was identified using standard molecular biology protocols by DNA amplification and sequencing of the ITS region. Genomic DNA extraction was performed using Intron's i-genomic BYF DNA Extraction Mini Kit according to the manufacturer's protocol. The nucleotide sequence of FCH061 was deposited in the GenBank database under accession number KU519426. The 18S rDNA sequence of this strain exhibited 99% identity (587/590) with that of Penicillium brasilianum (GenBank accession number AB455514).
Fermentation and extraction
The fungal strain was cultured on solid YPG media (5 g of yeast extract, 5 g of peptone, 10 g of glucose, 24.8 g of Instant Ocean, and 16 g of agar in 1 L of distilled water) for 7 days. An agar plug (1 cm × 1 cm) was inoculated for 7 days in a 250 mL flask that contained 100 mL of YPG media. Then, 10 mL of each culture was transferred to 2.8 L Fernbach flasks containing rice media (200 g of rice, 0.5 g of yeast extract, 0.5 g of peptone, and 12.4 g of Instant Ocean in 500 mL of distilled water). In total, 400 g of rice media was prepared and cultivated for 40 days at 28℃, with agitating once every week.
Isolation
The entire culture was macerated and extracted with EtOAc (1 L × 3). The solvent was evaporated in vacuo to afford a brown organic gum (8.2 g). The extract was separated by C18 reversed-phase vacuum flash chromatography using sequential mixtures of H2O and MeOH (six fractions of H2O-MeOH, gradient from 50:50 to 0:100), acetone, and finally EtOAc was the eluents. Based on the results of 1H NMR analysis, the fractions eluted with H2O-MeOH 20:80 (670 mg) and 10:90 (290 mg) were chosen for further separation. The fraction that eluted with H2O-MeOH (20:80) was separated by semi-preparative reversed-phase HPLC (YMC ODS-A column, 250 × 10 mm, 5 µm; H2O-MeCN, 58:42, 2.0 mL/min) to afford, in the order of elution, compounds 5, 6, 7, 9, 11, and 12. Purification of the ninth peak by reversed-phase HPLC (YMC-ODS-A column, 4.6 × 250 nm, 5 µm; H2O-MeOH, 42:58, 0.7 mL/min) provided compounds 1 and 2 as amorphous solids. The H2O-MeOH (10:90) fraction from vacuum flash chromatography was separated by semi-preparative reversed-phase HPLC (H2O-MeOH, 35:65, 2.0 mL/min), and afforded, in the order of elution, compounds 3, 4, 8, 10, 13, and 14. The overall isolated amounts of 1 – 14 were 6.9, 0.8, 24.6, 3.0, 8.8, 4.0, 3.8, 30.2, 10.0, 0.6, 16.4, 52.7, 24.6, and 3.8 mg, respectively.
Preaustinoid E (1)
white amorphous solid, [α]25D -26.9 (c 0.20, CHCl3); UV (MeOH) λmax (log ε) 211 (3.49) nm; 1H and 13C NMR: see Table 1; IR (ZnSe) νmax 3412, 2973, 2938, 1758, 1694 cm−1; HRFABMS, m/z 429.2275 [M+H]+ (calcd for C25H33O6, 429.2277).
-26.9 (c 0.20, CHCl3); UV (MeOH) λmax (log ε) 211 (3.49) nm; 1H and 13C NMR: see Table 1; IR (ZnSe) νmax 3412, 2973, 2938, 1758, 1694 cm−1; HRFABMS, m/z 429.2275 [M+H]+ (calcd for C25H33O6, 429.2277).
-26.9 (c 0.20, CHCl3); UV (MeOH) λmax (log ε) 211 (3.49) nm; 1H and 13C NMR: see Table 1; IR (ZnSe) νmax 3412, 2973, 2938, 1758, 1694 cm−1; HRFABMS, m/z 429.2275 [M+H]+ (calcd for C25H33O6, 429.2277).Preaustinoid F (2)
white amorphous solid, [α]25D -50.9 (c 0.20, CHCl3); UV (MeOH) λmax (log ε) 212 (3.83) nm; 1H and 13C NMR: see Table 1; IR (ZnSe) νmax 3120, 2973, 2931, 1755, 1687 cm−1; HRFABMS, m/z 429.2279 [M+H]+ (calcd for C25H33O6, 429.2277).
-50.9 (c 0.20, CHCl3); UV (MeOH) λmax (log ε) 212 (3.83) nm; 1H and 13C NMR: see Table 1; IR (ZnSe) νmax 3120, 2973, 2931, 1755, 1687 cm−1; HRFABMS, m/z 429.2279 [M+H]+ (calcd for C25H33O6, 429.2277).
-50.9 (c 0.20, CHCl3); UV (MeOH) λmax (log ε) 212 (3.83) nm; 1H and 13C NMR: see Table 1; IR (ZnSe) νmax 3120, 2973, 2931, 1755, 1687 cm−1; HRFABMS, m/z 429.2279 [M+H]+ (calcd for C25H33O6, 429.2277).Result and Discussion
The molecular formula of preaustinoid E (1) was deduced as C25H32O6 by HRFABMS analysis. The 13C NMR data of this compound showed signals of three carbonyl groups at δC 213.9, 171.7 and 167.6 which were indicative of a ketone and two ester carbonyls, respectively (Table 1). Signals of four olefinic carbons were also found at δC 155.3 (CH), 146.6 (C), 120.9 (CH), and 107.6 (CH2). Corresponding proton signals were found at δH 6.22 (1H, d, J = 12.2 Hz), 5.83 (1H, d, J = 12.2 Hz), 5.14 (1H, s), and 5.12 (1H, s) in the 1H NMR spectrum. The remaining signals in the 13C NMR data were attributed to six unprotonated, three methine, three methylene, and six methyl carbons. From the ten degrees of unsaturation inherent in the mass data, the 13C NMR data suggested 1 is a pentacyclic compound.
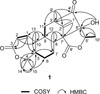
Given this information, the structure of compound 1 was determined by a combination of 2-D NMR experiments. First, all of the protons and their attached carbons were precisely matched by an HSQC experiment. Since the COSY data indicated only a small number of proton spin systems, the structure determination was mainly accomplished by the HMBC experiments (Fig. 2). That is, the significant differentiation of the COSY signals corresponding to the double bond (δC 155.3, δH 6.22; δC 120.9, δH 5.83) suggested it was in conjugation with an electron-withdrawing group, which was confirmed by its long-range coupling with a carbonyl carbon (δC 167.6) in the HMBC data (C-1-C-3). In addition, the HMBC correlations of two methyl groups (δC 32.3, δH 1.40; δC 26.6, δH 1.42) with an unprotonated carbon (δC 85.5) readily defined an isopropyl group (C-4, C-14 and C-15). Additional HMBC correlations of these methyl groups suggested an adjacent methine carbon (C-5; δC 56.0). Then, the connection of this moiety with the pre-defined conjugated carbonyl group via an unprotonated carbon (C-10; δC 43.7) was also constructed by a series of HMBC correlations including a methyl proton (H3-13; δH 1.16); H-1/C-5, H-2/C-10, and H3-13/C-1, C-5, C-10. The diagnostic shielding (δC 85.5) of the C-4 carbon suggested an ester bridge with the C-3 carbonyl carbon constructing a 7-membered lactone (ring A; C-1-C-5, C-10, and C-13), which was confirmed by interpretation of the mass data (discussed later).
The COSY data revealed a direct connection between the C-5 methine and the ethylene group at C-6 and C-7, which was extended by a HMBC correlation with an isolated methyl group (C-12; δC 18.2, δH 1.29). Subsequently, the HMBC correlations of H3-12 and H3-13 with a common methine (C-9; δC 47.8, δH 2.15) constructed a 6-membered ring with two methyl substituents (ring B; C-5-C-10, C-12 and C-13). Similarly, another 6-membered ring (ring C; C-8, C-9, C-11, C-2′, C-3′, and C-7′) as well as attachment of an exomethylene (C-1′) and a methyl group (C-9′) to this ring was constructed by a COSY-derived proton spin system (H-9-H2-11) coupled with the HMBC correlations of key protons with neighboring carbons: H3-12/C-7′; H2-1′/C-3′ and C-7′; and H3-9′/C-11, C-2′ and C-3′. An additional HMBC correlation with H3-9′ placed a ketone carbon at the neighboring position (C-4′; δC 213.9), and this fragment was extended by long-range couplings of a hydroxyl proton (δH 3.05) with this ketone and an unprotonated carbon (C-6′; δC 90.6). The latter carbon was also connected to a COSY-derived methyl methine moiety (C-5′ and C-10′) by the HMBC correlations of H-5′/C-6′ and H3-10′/C-6′.
The proton-proton and proton-carbon based 2-D NMR analyses indicated an ester carbonyl carbon (δC 171.7) and six open ends (C-3, C-4, C-5′, C-6′, and C-7′ (two ends)), and of these, the latter accounts for three of the degrees of unsaturation inherent in the mass data (Fig. 2). The significant shielding of the C-5′ methine (δC 75.6, δH 4.23) was indicative of an ester conjugated with a carbonyl group (C-8′). Therefore, the only plausible connections of these fragments in the eastern portion of 1 were those between C-6′ and C-7′ and between C-7′ and C-8′, establishing a ketone-bearing 5-membered ring (ring D; C-2′-C-4′, C-6′, and C-7′) and a 5-membered methyl lactone (ring E; C-5′-C-8′ and C-10′), respectively. Similarly, an ester bridge was also placed between C-3 and C-4 to form a 7-membered lactone (ring A). These interpretations were supported by a literature study, and the NMR data of these moieties were in good agreement with reported values.12131415
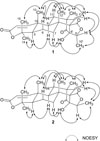
Preaustinoid E (1) possessed several stereocenters mostly at the ring junctions. Accordingly, the configurations at these positions were assigned by conspicuous cross-peaks among the bridgehead methyl protons and neighboring protons in the NOESY data (Fig. 3). That is, H3-12, H3-13, and H3-15 were axially oriented and cofacial by their characteristic NOESY cross-peaks: H-6β/H3-12, H-6β/H3-13, H-11β/H3-12, H3-12/H3-13, and H3-13/H3-15. In contrast, the H-5 and H-9 methines were located on the opposite face, indicating of trans A/B and B/C ring junctures, by the NOESY correlations of H-5/H-9 and H-5/H3-14.
The configurations of the remaining portion of 1 were also assigned based on the NOESY data. That is, the chair type conformation of the C-ring as well as the equatorial orientation of the C-9′ methyl group was derived from the spatial proximity between the C-12 methyl and the C-1′ exomethylene group: H-11β/H3-9′, H3-12/H2-1′ (δH 5.14), and H3-9′/H2-1′ (δH 5.12). In addition, the α- and axial orientations of the D/E ring juncture were defined by the cross-peaks of 6′-OH with those on the A-C ring plane: H-7α/6′-OH and H-9/6′-OH. Similarly, a cis orientation was found between 6′-OH and 10′-CH3 on the E ring: 6′-OH/H3-10′. These assignments were supported by the NOESY cross-peaks between H2-1′ and H-5′, which could be due to not only an axial orientation of the groups at the D/E ring junction and β-orientation of H-5′ relative to the E-ring but also the β-orientation of the C-8′ carbonyl carbon at C-7′ (Fig. 3). Overall, the relative configuration of 1 was assigned as 5R*, 8R*, 9R*, 10R*, 3′S*, 5′S*, 6′S*, and 7′S*, which was in good accordance with that of similar compounds.131416 Thus, the structure of preaustinoid E (1) was determined to be a new austin-type meroterpenoid.
The molecular formula of preaustinoid F (2) was established to be C25H32O6, the same as 1, by HRFABMS analysis. The spectroscopic data of this compound were very similar to those of 1, suggesting their structures were closely related. A combination of 1-D and 2-D NMR analyses confirmed their similarity and indicated 2 and 1 had the same planar structure. However, detailed examination of the 1H and 13C NMR data revealed significant differences in the chemical shifts of the protons and carbons at the C-5′ methine, C-10′ methyl and 6′-OH groups, suggesting they were C-5′ and/or C-6′ epimers (Table 1). This interpretation was confirmed by the NOESY data of 2 in which opposite cross-peaks were found for H-5′ and H3-10′: H2-1′/H3-10′ and H-5′/6′-OH (Fig. 3). Thus, the structure of preaustinoid F (2) was defined as the C-5′ epimer of preaustinoid E (1).
In addition to compounds 1 and 2, extensive separation of the moderately polar chromatographic fractions allowed us to isolate several austin-type meroterpenoids. A combined of spectroscopic analyses of these congeners identified twelve known compounds, preaustinoid A2 (3), preaustinoid D (4), dehydroaustinol (5), dehydroaustin (6), actoxydehydroaustin (7), isoaustinone (8), 11β-acetoxyisoaustinone (9), (5′R)-isoaustinone (10), austin (11), neoaustin (12), austinoneol A (13), and verruculogen (14), the only prenylated diketopiperazine (Fig. 1). All the spectroscopic data for these compounds were in good accordance with those in the literature.512131415161718192021
Austin-type meroterpenoids have attracted significant biosynthetic interests.789 Unfortunately, these compounds failed to exhibit significant bioactivities, and only insecticidal and weak bacteriostatic activities against E. coli have been reported.1622 In our measurement, these compounds were also inactive against a variety of human cancer cell-lines (IC50 > 50 µM) and human pathogenic bacterial and fungal strains (MIC > 128 µM). In addition, none of these meroterpenoids exhibited any positive results (IC50 > 100 µM) in tests of selected enzyme-inhibition (isocitrate lyase, Na+/K+-ATPase, and sortase A) activities.

 XML Download
XML Download