

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
The innate immune response is a crucial component of the first-line defense system against a variety of invading pathogens (12). Macrophages are principal innate immune cell types that participate in recognition, signaling, digestion, antigen presentation, and effector functions in the infections to a variety of pathogens (23). Toll-like receptors (TLRs) are one of the best characterized innate immune receptors that can sense a variety of pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs) during bacterial and viral infections (145). In macrophages, TLR engagement by various PAMPs and DAMPs is importantly involved in the activation of intracellular signaling cascades that result in the activation of effector mechanisms and pathways to eradicate the invading pathogens (67). The activation of TLR responses triggers the sequential activation of signaling proteins and adaptors, effector enzymes through cooperative assembly mechanisms that elicit the innate immune responses (4578). The effector pathways of innate immune system consist of the expression of proinflammatory cytokines and chemokines, generation of reactive oxygen species (ROS) and nitrogen species, and production of antimicrobial proteins (79).
Autophagy is an intracellular catabolic process that can promote the destruction of invading pathogens in the autolysosomes (10). Accumulating evidence suggest that autophagy acts as a critical effector system in the host cells when they are invaded by pathogens (111213). Therefore, numerous bacteria and virus have evolved multiple strategies to exploit or evade from host defensive autophagy (101113). Indeed, TLR signaling can activate autophagy and lysosomal function, and LC3-associated phagocytosis (LAP) when combined with phagocytosis during infection, which contribute to innate effector function (1415). Since there are numerous studies for the role of autophagy in the regulation of inflammation (161718), it may contribute to control excessive inflammatory responses during pathogenic infection. We summarize the TLR-mediated activation of innate effector mechanisms and particularly focus on the role of autophagy during intracellular bacterial infection.
Overview of TLR-mediated Intracellular Signaling in Macrophages
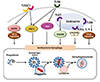
TLRs are the distinct pattern recognition receptor (PRR) members that include 10 and 12 family members in human and mice, respectively (8). TLR has a three-domain structure, leucine-rich repeats (LRRs) for the recognition of PAMPs, a transmembrane region, and an intracellular Toll/IL-1 receptor (TIR) domain for the signaling activation. In addition, TLRs are localized either in the plasma membrane (TLR1, TLR2, TLR4-6, and TLR10) or endosomal membranes (TLR3, TLR7-9, TLR11-13) (19). In addition, recent studies identified the molecules, such as uncoordinated 93 homolog B1 (UNC93B1) and leucine-rich repeat containing protein (LRRC) 59, for mediating endosomal TLR trafficking from ER to intracellular compartments (2021).
Upon the LRR engagement of TLRs by various PAMPs or DAMPs, TLRs trigger the recruitment of TIR domain-containing adaptors such as myeloid differentiation primary response protein 88 (MyD88) and TIR domain-containing adaptor protein inducing IFNβ (TRIF), TIR domain-containing adaptor protein (TIRAP)/MyD88 adapter-like protein (Mal), or TRIF-related adaptor molecule (TRAM). MyD88 is recruited by all TLR molecules, whereas TRIF is utilized by TLR3 and 4. The recruitment of MyD88 leads to the phosphorylation and activation of IL-1 receptor-associated kinase (IRAK) kinase family members including IRAK4 and IRAK1, which associates with E3 ubiquitin ligase TNF receptor associated factor (TRAF) 6 to activate the “master” TAK1 protein kinase complex (2223). Upon activation, TAK1 induces the activation of both NF-κB and mitogen-activated protein kinase (MAPK) pathways (ERK1/2, p38, and JNK) for further activation of transcriptional factors including AP-1 family members to produce proinflammatory cytokines including tumor necrosis factor (TNF)-α, interleukin (IL)-6, and various chemokines (824). In addition, the other adaptor TRIF interacts with TRAF6 and TRAF3, to facilitate the activation of TAK1 complex and TBK1 pathways, respectively. TRIF-dependent TRAF3 activation leads to the activation of type I interferon responses through phosphorylation of transcriptional factor IRF3 (82425).
During this sophisticated signaling activation, ubiquitination plays a key orchestrating role for controlling innate immune responses and inflammation through post-translational modification of target proteins (26). Both TRAF6 and TRAF3 are well-known and essential ubiquitin ligases that induce polyubiquitylation of target proteins to recruit for the activation or degradation during TLR signaling. Numerous studies identified crucial E3 ubiquitin ligases that play key regulatory roles in TLR signaling (27). For example, the Pellino family E3 ubiquitin ligases are involved in K63-linked polyubiquitylation of K63-linked polyubiquitination of IRAK1, TBK1, TAK1, receptor-interacting protein 1 (RIP1; also known as RIPK1) in TLR signaling pathways (272829). A recent study showed that the ubiquitin ligase RNF19A is importantly involved in the K48-linked ubiquitination and degradation of TRAF6, thereby attenuating TLR signaling (30). Figure 1 summarizes an overview of TLR-induced intracellular signaling pathways in innate immune cells. Deciphering the detailed mechanisms by which TLR signaling is regulated in macrophages may suggest future strategies to enhance host innate defense and prevent excessive inflammation.
Effector molecules in TLR-mediated innate immune responses
Upon TLR activation, the intracellular signal transduction via numerous adaptors/enzymes leads to the activation of multiple effector mechanisms and pathways. Here we will review three major innate immune pathways, antimicrobial proteins, mitochondrial ROS, and auutophagy, in terms of TLR activation.
Antimicrobial proteins in TLR responses
TLR-mediated innate immune signaling ultimately activates the exprssion of numerous antimicrobial peptides (AMPs) which are multifunctional molecules that kill pathogens, play immunomodulatory roles, and inflammation, in various settings of infection and inflammation (313233). Here we discuss two major types of AMPs, defensins and cathelicidins, in terms of TLR signaling.
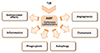
Both defensins and cathelicidins are principal AMPs that can be synthesized in various cell types including innate immune cells, such as epithelial cells and macrophages. Previous studies suggest that TLR signaling leads to the differential profiling of AMP production depending on the specific cell or tissue types. Recent studies showed that the TLR4-mediated increase of β-defensin 2 expression in mesenchymal stem cells elicited antibacterial effects and down-regulated inflammatory responses during Escherichia coli-induced pneumonia (34). TLR stimulation contributed to the enhancement of host defense through the induction of nondefensin proteins, but not defensin-family proteins, to increase antibacterial activity in the intestinal mucosa after antibiotics treatment (35). In addition, there is a synergistic activation of antimicrobial peptide production when both TLRs and nucleotiden binding oligomerization domain 1 and 2 (NOD1 and NOD2) are stimulated together (36).
Recent studies provided the evidence for the opposite regulation of AMP in TLR-mediated immune responses that reshape the host defense and inflammation (31). For example, cathelicidins negatively regulate macrophage activation and inflammation through binding lipoproteins and LPS (37). LL-37, a C-terminal portion of human cathelicidin antimicrobial protein (hCAP18) (38), is known to decrease TLR4 signaling, but enhance TLR3 signaling (3940). Indeed, LL-37 can bind to various agonists of TLRs, and affect TLR-mediated signaling and bacterial phagocytosis (394041). In addition, there is a synergistic interaction between AMP and TLR signaling in the activation of cytokine production. Previous studies showed that human β-defensin-2 and -3 led to a synergistically increased production of inflammatory cytokines and chemokines in response to TLR ligands, through ATP release (42). Another study showed that human β-defensin 3 had an inhibitory function in the TLR4-mediated transcriptional activation of cathelicidon of proinflammatory genes in macrophages (43). Although it is not clear in terms of TLR signaling, the function of cathelicidins has been reported in the modulation of angiogenesis, presumably linked to tumorigenesis and inflammation (4445). The interaction of cathelicidins and defensins with microenvironment results in multifunctional roles in infection and inflammation (Fig. 2).
Importantly, TLR2/1 activation led to an induction of vitamin D signaling pathway and LL-37 production to enhance intracellular killing effects in human monocytes/macrophages against infection with Mycobacterium tuberculosis (Mtb), a major pathogen of human tuberculosis (TB) (46). In addition, TLR2/1 activation stimulated the induction of defensin-β4, which depends on IL-1β production in human monocytes (47). Moreover, TLR2-mediated hippo (mammalian sterile 20-like 1 and 2 kinases, MST1/2, inmammals) signaling is required for the paracrine activation of antimicrobial peptide β-defensins through CXCL1 and CXCL2 secretion during Mtb infection (48). These data suggest that TLR signaling cooperates with other signaling pathways for the induction of AMP to contribute to TLR-mediated antimicrobial host defense.
TLR and mitochondrial ROS
Recent studies provided evidence that mitochondria are crucial organelles to regulate innate immune responses to various DAMP and PAMP signals (49). In macrophages, activation of TLR1, 2, and 4 are associated with the increased production of mitochondrial ROS, which are critically involved in bactericidal activities and the recruitment of mitochondria to bacterial phagosomes (50). This response is mediated by the translocation of the TRAF6 to the mitochondria, and the TRAF6 interaction with ECSIT (evolutionary conserved signaling intermediate in Toll pathways), a protein that is esential for the respiratory chain assembly and increased mitochondrial ROS generation (50). A more recent paper revealed the mechanisms for mitochondrial trafficking and juxtaposition to bacterial phagosomes are mediated by the kinases Mst1 and Mst2 via the activation of the Rho family GTPase Rac (51). The GTPase Rac activation is required for the TLR-mediated interaction of TRAF6-ECSIT, thereby recruiting mitochondria to phagosomes and subsequent killing of microbes (51). Interestingly, peroxiredoxin-6, an antioxidant enzyme of peroxiredoxin family, is required for the inhibition of mitochondrial ROS production through interruption of the TRAF6-ECSIT complex formation in response to TLR4 (52). In addition, a recent paper showed another function of mitochondrial ROS production that is required for the antigen cross-presentation capacity of plasmacytoid dendritic cells after TLR stimulation (53).
Autophagy: Effector Pathway in innate immune responses by TLR signaling
Autophagy is a lysosomal degradation pathway and acts as an innate immune effector because it mediates the clearance of intracellular microbes (5455). In addition, autophagy plays an important role in various aspects of immune responses, including antigen presentation, regulation of cytokine production, and lymphocyte homeostasis (55). Here we briefly discuss the overview of autophagy, TLR-induced autophagy and its consequences during infection, and the mechanisms for crosstalk between TLR signaling and autophagy process.
Overview of autophagy and xenophagy
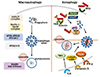
Autophagy process is required for intracellular maintenance of homeostasis during various stress signals including nutrient starvation and pathogenic invasion (5657). The autophagic process is divided at least three steps: initiation, elongation, and maturation. In the initiation step, cargos are surrounded by a cup-shaped double-membrane structure, phagophore, through the action of ULK1/2 complex (ULK1/2-Atg13-Atg101-FIP200) and Class III PI3K-Beclin-1 complex (Beclin 1-ATG14L-Vps34-p150). The phagophore structure expands to autophagosomes, through two ubiquitin-like conjugation systems containing core autophagy proteins including Atg8/LC3 and Atg5-Atg12-Atg16L1 (56). In the autophagosome maturation step, the autophagosomal structures can be fused with endolysosomal vesicles to form degradative autolysosomes through several proteins including SNAREs, ATG8 family members, and Rab GTPases (58). Since autophagy is essential in the maintenance of house-keeping function and homeostasis, its dysfunction and altered regulation are associated with numerous diseases (59).
Although autophagy was considered to be a nonspecific response, it is now being clear that autophagy can target specific organelles or substances including pathogens, i.e., xenophagy (57). Numerous autophagic adaptors including p62, NDP52, and optineurin, play an important role in the activation of selective autophagy activation (606162). Since the autophagic adaptors have both domains, i.e., LC3-interacting region and ubiquitin-binding domains, to connect ubiquitinylated cargos to autophagic machinery (6061). Numerous pathogens have evolved different mechanisms to escape or exploit autophagy/xenophagy to get an advantage for the survival of pathogens (63). Here we briefly introduce xenophagy against Mtb infection, because xenophagy has been widely studied in Mtb infection.
Mtb can reside in the phagosomes in macrophages, but access into the cytosols through ESX-1 system (64). Mtb and their DNA can be recognized by cytosolic sensor c-GAS, and subsequently ubiquinylated by Parkin and Smurf1 (656667). Xenophagy against Mtb is mediated through autophagic adaptors including p62 and NDP52 (64). In addition, TRIM and Galectin family proteins cooperate the recognition of damaged phagosomes, thereby the core autophagy proteins can be targeted to activate xenophagy process (6869). In this process, the mycobactericidal activity is presumably due to the formation of neo-antimicrobial peptides, which are delivered from numerous bulk ubiquitinated proteins during autophagic process, through the action of p62 (7071). In human monocytes/macrophages, vitamin D3-induced autophagy activation was essentially required for antimicrobial responses through induction of cathelicidins (727374). In addition, IRGM, a human immunity-related GTPase, induces xenophagy through interaction with ULK1 and Beclin 1, to enhance the formation of autophagy initiation complex and antimicrobial functions (75). Figure 3 summarizes a brief overview of autophagy and xenophagy during Mtb infection.
Although there are a large body of evidence that autophagy/xenophagy promotes antimicrobial responses to Mtb infection, recent studies argued about the function of autophagy in vivo. Genetic deletion models of autophagy genes in myeloid lineage of the mice did not exhibit protective effects against Mtb infection in vivo, except Atg5 that showed an autophagy-independent, and controlling neutrophil-mediated harmful inflammation to the host (76). Despite this, autophagy may contribute to the protective immune responses and improvement of antigen-presentation, thus being a promising target for the development of new vaccines and therapeutics (77).
TLR-mediated autopagy regulation
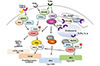
TLR signaling activation in innate immune cells results in various effector mechanisms/pathways including autophagy process, during pathogenic infection (78). Previous studies showed that TLR signaling activation led to the induction of autophagy in macrophages (7980). Among TLRs, TLR7 activation showed strong effects upon the enhancement of autophagy leading to the elimination of intracelular microbes in macrophages (79). TLR7-mediated autophagy depended on MyD88 expression (79). In addition, TLR4-induced autophagy was mediated through TRIF-dependent, but MyD88-independent, pathway (81). Interestingly, both TLR4- and TLR7-mediated autophagy activation resulted in the increased colocalization of mycobacterial phagosomes and autophagosomes, indicating a promotion of phagosomal maturation (8081).
In human monocytes/macrophages, TLR2/1 activation activated AMPK-dependent functional vitamin D signaling activation, which leads to the induction of autophagy activation and antimicrobial responses (82). Another study showed that treatment of murine macrophages with TLR4/LPS or TLR3/poly(I:C) resulted in the elimination of intracellular mycobacteria through autophagy activation (83). Either stimulation of TLR2 or TLR4 enhanced the expression of a serine protease inhibitor plasminogen activator inhibitor type 2 (PAI-2), which stabilized Beclin 1 to activate autophagy pathway to suppress NLRP3 activation (84). Thus TLR-mediated autophagy activation may contribute to cell-autonomous antimicrobial defense and controlling excessive inflammation. In other aspect, TLR signaling plays a negative regulatory role in autophagy activation in different context. Recent studies showed that TLR4/LPS stimulation inhibited autophagy in microglial cells through negative regulation of FOXO3 at downstream of PI3K pathway (85). In Leishmania infection, TLR3, 7, 9, as well as UNC93B1, a chaperone for trafficking of nucleic-sensing TLRs to endolysosomes, are importantly required for the activation of anti-microbial autophagy and controlling parasite replication (86).
TLR8 stimulation robustly activated the expression of genes involved in vitamin D signaling, which promoted vitamin D-cathelicidin-dependent autophagy and antimicrobial responses against human immunodeficiency virus infection in human macrophages (87). Thus TLR signaling may adopt distinct mechanisms and effects upon autophagy in the different context of manner. A brief overview that TLR signaling regulates antibacterial autophagy is shown in Figure 4. In addition, TLR4 stimulation led to an induction of LC3-positive structures, which are independent of canonical macroautophagy and contain p62 as a component (88). The formation of p62-positive selective autophagosome was mediated through Nrf2 and TLR4-MyD88 signaling pathways (88). These data suggest that TLR-mediated innate immune and ROS pathways converge to autophagy activation that contribute to promote host defense during infection.
Crosstalk between TLR signaling molecules and autophagy proteins
There are several evidence that the autophagy pathway regulates TLR-mediated innate immune responses, suggesting there is a complex link between TLR responses and autophagy. For instance the formation of immunoamphisome in plasmacytoid dendritic cells enhanced TLR7-mediated responses and antigen presentation on both MHC class I and class II molecules (89). In intestinal epithelial cells, TLR-induced autophagy and Atg7 expression are required for IL-8 production, suggesting a role for autophagy in intestinal innate immune responses (90). In contrast, suppression of autophagy pathway by either pharmacological inhibitors of autophagy or RNA interference amplified the IL-23 production in macrophages and dendritic cells in response to TLR activation (91).
A large body of evidence showed that there is intimate connection between TLR signaling-related molecules and autophagy-related proteins. Earlier studies showed that TLR family adaptors MyD88 and TRIF were associated with Beclin-1, a key autophagy-related protein in the autophagosome formation (92). It was also reported that TRAF6 was essentially involved in the K63-linked polyubiquitination of Beclin-1 to induce TLR4-mediated autophagy in macrophages (9394). In addition, a well-known deubiqutinase A20 suppressed the K63-linked polyubiquitination of Beclin-1 to inhibit the autophagosome formation (9394). During Pseudomonas aeruginosa infection, NLRC4 inflammasome-dependent caspase-1 activation led to a cleavage of TRIF, thus attenuating autophagy activation and type I interferon expression (95). In vivo infection model with P. aeruginosa showed that TRIF was required for the enhancement of antibacterial autophagy and clearance of bacteria (95). A recent study showed that the TLR adaptor molecule Mal (encoded by TIRAP) through IFN-γ receptor signaling, which in turn activated autophagy and increased the protection from M. tuberculosis infection (96). This study is important for the potential explanation of genetic variant of Mal (S180L polymorphism; murine equivalent S200L) in the increased susceptibility to TB (96). Another recent study revealed that TRIF degradation, which was mediated through selective autophagic pathway via the E3 ligase TRIM32 and the autophagic adaptor TAX1BP1, negatively regulated TLR3/4-induced type I interferon and proinflammatory immune responses (97).
There are several reports that autophagy proteins are involved in innate immune responses associated with TLR signaling. In mycobacterial infection models, the autophagy protein DNA damage-regulated autophagy modulator (DRAM) 1 expression was dependent on MyD88- and NF-κB-mediated innate immune signaling in human macrophages and zebrafish embryos (98). Moreover, the autophagy inhibitor Rubicon played a role as a feedback regulator of CARD9, BCL10, and MALT1 (CBM complex)-mediated innate immune signaling (99). Rubicon functioned in the disassembly of CBM complex formation and inhibited cytokine production (99). In addition, TLR2 activation led to an interaction of Rubicon with the p22phox of NADPH-oxidase complex for the enhancement of antimicrobial effects through induction of ROS and inflammatory cytokine generation (100). A recent study showed that Rubicon plays an essential role for antiviral type I interferon responses through interaction with interferon regulatory factor (IRF) 3 and inhibition of IRF3 dimerization (101). Together, these studies indicate an essential role for TLR signaling adaptors in the regulation of the autophagy protein function or vice versa.
Concluding remarks
Considerable progress has been made in elucidating the mechanisms for TLR-induced signaling in innate immunity. For innate immune cells such as macrophages, the activation of TLR-mediated innate immune signaling leads to the effector pathways including cytokine generation, reactive nitrogen and oxygen species, production of antimicrobial proteins, autophagy, etc. Although a great advance has been done in the unveiling the signaling mechanisms by which TLR activation leads to the innate immune responses, many questions remained in the functional identification of positive and negative regulators in TLR-mediated signaling pathways.
In addition to providing a signaling map of TLR pathway, future studies are warranted to investigate the immunomodulatory functions of diverse antimicrobial proteins in innate immune responses. Numerous efforts support a role for mitochondrial ROS in the antimicrobial responses against pathogens. However, dysregulation of ROS generation could amplify chronic inflammation during infection. Thus it remains to be seen whether TLR-mediated autophagy or other cellular pathway is required for the coordinated regulation of innate effector responses.
The information on the function of autophagy/xenophagy is emerging in a variety of infection; one example would be Mtb-induced host defensive responses. Accumulating evidence suggests that TLR signaling activates autophagy, which affects TLR-mediated innate immune responses during infection. Understanding the molecular mechanisms for biological connections between autophagy and TLR signaling is a challenge for the possibility of novel therapeutics against infection and inflammation.
 XML Download
XML Download