

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Castleman disease (CD), characterized as either unicentric CD (UCD) or multicentric CD (MCD), is an uncommon lymphoproliferative disorder with unclear underlying mechanisms [1]. It most frequently develops at the mediastinum (67%) and less commonly at the retroperitoneal cavity (12%) [2]. Transplantation-related CDs have been reported in the literature. There was a case report on a patient with mediastinal mass associated with paraneoplastic pemphigus and bronchiolitis obliterans who was diagnosed with CD and thus required pulmonary transplantation [3]. Several reports have demonstrated that human herpesvirus-8 (HHV-8) and Epstein-Barr virus (EBV)-associated monotypic large B-cell lymphoproliferative disorders are associated with mixed-variant CD after kidney transplantation (KTP). A case of CD after KTP was also reportedly resolved after graft nephrectomy [4]. In addition, a Japanese KTP recipient recovering from end-stage renal failure due to MCD was also reported [5]. Herein, we report a Korean case of CD that improved after KTP.
CASE REPORT
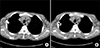
A 56-year-old man with hypertensive nephropathy was undergoing hemodialysis (HD) since 2008. On December 28, 2011, the fourth year of HD treatment, he complained of abdominal discomfort. The physical examination showed no specific findings. Meanwhile, his laboratory findings showed a serum creatinine (Cr) level of 8.9 mg/dL, blood urea nitrogen of 49.1 mg/dL, C-reactive protein (CRP) of 9.85 mg/dL, lactate dehydrogenase of 115 IU/L, hemoglobin of 9.0 g/dL, platelet count of 262,000/mm3, and serum albumin of 3.4 g/dL. Chest X-ray results did not show any specific findings. An abdominopelvic computed tomography (CT) scan revealed multiple, variable-sized, and homogeneously enhanced nodular densities in the mesenteric, retrocaval, paraaortic, and bilateral inguinal regions (sizes: up to 15 mm in the left paraaortic and 22 mm in the left inguinal region) (Fig. 1). Subsequently, a left inguinal lymph node excisional biopsy was performed on January 9, 2012 (Fig. 2). The specimen consisted of a soft-tissue mass measuring 4.5×4×1.5 cm. The cut surface revealed enlarged multiple lymph nodes (largest node size, 4×2 cm cross-sectionally). The tissue was submitted for light microscopy, and paraffin sections were stained with hematoxylin and eosin [6]. Pathologically, the mass was diagnosed as CD, plasma-cell type. Therefore, HD was performed, which alleviated his chief complaint after the supportive care. No CD treatment was performed, as no evidence of relapse was observed after symptom recovery. The patient underwent deceased-donor KTP on March 9, 2017. The preoperative abdominopelvic CT scan revealed persistent lymphadenopathy. This represented no significant difference, which is inconsistent with that of the previous CT scan findings (Fig. 3A). On chest CT scan, bilateral hilar and mediastinal lymph nodes and bilateral axillary lymph nodes, previously unverified, were identified (Fig. 4A).
The patient's panel reactive antibody results were 19% in class I and 9% in class II, but no donor-specific antibody was detected. Human leukocyte antigen mismatch 4/6 (recipient: A 2/24, B 35/54, DR 14/15; donor: A 2/24, B 27/44, DR 1/7) was observed. Postoperative immunosuppressive therapy consisted of a triple-drug regimen: tacrolimus at a starting dose of 0.075 mg/kg/day and adjusted when the blood trough level was >10 ng/mL; mycophenolate mofetil 1,500 mg/day, and a 2-month tapered dose of prednisolone from 250 to 5 mg/day. Basiliximab 20 mg was administered on the operation day and 4 days after KTP.
Intraoperatively, excisional biopsy was simultaneously performed on the lymph nodes located in the external iliac area. The specimens included two enlarged lymph nodes measuring 3×2×0.9 cm and 3.5×1.5×1 cm. The histologic examination revealed plasma-cell-type CD showing histopathological findings similar to the previous biopsy results and no malignant progression. EBV immunoglobulin G (IgG)-positive and IgM-negative findings were also assessed, and HHV-8-negative results were found. Repeat CT scan was performed 3 weeks postoperatively, on March 31, 2017. Compared with the CT scan taken during KTP, the lymph nodes decreased in size (Figs. 3B, 4B). One year after KTP, the patient has normal graft function (serum Cr of approximately 1.4 mg/dL) without clinical or pathological evidence of rejection. In addition, surveillance with radiologic (Fig. 3C) and hematologic follow-up demonstrated no evidence of CD aggravation.
DISCUSSION
CD is classified into at least three distinct disorders based on the number of regions of enlarged lymph nodes with characteristic histopathologic features and presence/absence of HHV-8 (also known as Kaposi-sarcoma-associated herpesvirus) infection [789]. UCD involves one or more enlarged lymph nodes in a single region of the body, whereas MCD involves multiple regions of lymphadenopathy. MCD is categorized based on HHV-8. HHV-8-negative idiopathic MCD (iMCD) has at least three subgroups: polyneuropathy, organomegaly, endocrinopathy, monoclonal protein, skin changes (POEMS)-associated; thrombocytopenia, anasarca, fever, reticulin fibrosis, and organomegaly (TAFRO) syndrome; and not otherwise specified (NOS) [1011]. The patient was classified as having HHV-8-negative iMCD owing to lymphadenopathy in multiple regions and HHV-8-negative status. Moreover, as he did not have POEMS syndrome or the TAFRO subtype, he was classified as having iMCD-NOS.
The etiology of iMCD is unknown; however, possible mechanisms have been suggested to include autoimmune, autoinflammatory, neoplastic, and infectious processes [1213]. Interleukin (IL)-6 is an essential and sufficient factor for its symptomatology, histopathology, and pathogenesis. IL-6 is a multifunctional cytokine characterized by plasmacytosis, hypergammaglobulinemia, thrombocytosis, acutephase protein production, and activation of macrophages and T cells [14]. In addition to IL-6, vascular endothelial growth factor IL-1 is also involved in the etiology of iMCD [1516]. In this patient, the autoantibodies were negative, and none of the clinical pathological features of autoimmune disease were found. In fact, notwithstanding the fact that these features are typically observed in lymphoma, they were not evident in our patient. However, no infections caused by pathogens other than HHV-8 were identified either. In addition, the mechanism of germline mutations in genes regulating the inflammation was not confirmed. Therefore, either an infectious or an autoinflammatory mechanism was suggested to be involved.
The following two major criteria must be satisfied to diagnose iMCD: enlarged lymph nodes (≥1 cm in the short-axis diameter), ≥2 lymph-node stations, and histopathologic lymph-node features [17]. The iMCD spectrum is characterized by regressed/atrophic/atretic germinal centers, follicular dendritic cell prominence, vascularity, polytypic plasmacytosis in the interfollicular space, and hyperplastic germinal centers. iMCD is also diagnosed when at least two of the 11 minor criteria were found, with at least one of them as a laboratory criterion. The minor criteria consist of laboratory and clinical data. The minor laboratory criteria include elevated CRP or erythrocyte sedimentation rate, anemia, thrombocytopenia or thrombocytosis, hypoalbuminemia, renal dysfunction or proteinuria, and polyclonal hypergammaglobulinemia. The minor clinical criteria include constitutional symptoms (night sweats, fever, weight loss, or fatigue), enlarged spleen and/or liver, fluid accumulation, eruptive cherry hemangiomatosis or violaceous papules, and lymphocytic interstitial pneumonitis. However, even if these criteria are all met, various infection-related disorders, autoimmune/autoinflammatory diseases, and malignant/lymphoproliferative disorders that may be confused as iMCD should still be excluded.
The natural history of HHV-8-negative iMCD also varies. The prognosis of untreated MCD is poor. The 5-year overall survival ranges from 55 % to 77% [1819]. However, the indolent form, rather than the rapidly progressive form, does not seem to deteriorate with time. Disease severity should be assessed by determining the direction of iMCD treatment [20]. In non-severe diseases without life-threatening organ failure, an anti-IL-6 monoclonal antibody, siltuximab, or an anti-IL-6 receptor monoclonal antibody, tocilizumab, is the primary iMCD treatment. Siltuximab (11 mg/kg intravenous infusion every 3 weeks for 12 weeks) has been evaluated in a double-blind, placebo-controlled, randomized study using a control arm of best supportive care with up to 60 mg of prednisone. The combined long-term symptomatic and tumor response was 34% [21]. A single-arm study on 28 Japanese patients receiving tocilizumab (8 mg/kg intravenously every 2 weeks for 16 weeks) demonstrated a high response rate in terms of symptoms, laboratory parameters, and reduction of lymphadenopathy [22]. Even if a sufficient response after treatment is detected, the IL-6 inhibitor should be maintained to prevent symptom recurrence when the treatment is stopped (remission is defined as the absence of abnormalities on physical examinations and laboratory or imaging studies). In response to insufficient response or progressive organ failure during response access, systemic chemotherapy with or without an immunomodulator/immunosuppressant or rituximab may be considered [23]. Patients with severe iMCD who have marked organ dysfunction, poor performance status, and/or require critical care should be promptly started on a high-dose steroid regimen (e.g., methylprednisolone 500 mg daily) together with siltuximab or tocilizumab. However, POEMS-associated iMCD does not respond to IL-6 inhibitors; therefore, hematopoietic cell transplantation and radiation treatment can be considered [20]. The present patient was not treated with IL-6 inhibitors during KTP. However, glucocorticoids used during KTP relieved the symptoms by inhibiting hypercytokinemia [23], and tacrolimus prevented CD deterioration by inhibiting T helper 1 cell inflammation, which is considered as one of the CD pathologies [24].
Active malignancy is an absolute contraindication for KTP. Patients with posttransplant lymphoproliferative disorder (PTLD) should not receive a second transplant until at least 2 years after a successful treatment [25]. A lymphoproliferative disorder such as CD may be found as a posttransplant complication. Determining an indication for KTP is difficult in a potentially systemic disease such as MCD. However, some reports demonstrated that CD does not deteriorate after KTP. A case of hyaline vascular-type CD incidentally found during KTP showed no major complications at the 3-year follow-up [26]. There is also a case report of CD recovery after lung transplantation [27]. In our case, either active CD or PTLD was rarely detected, and no clear contraindication was evident. If symptoms of MCD are maintained without deterioration, as in the present case, transplantations can be performed successfully.
In conclusion, an iMCD patient who underwent HD for end-stage renal disease was successfully managed with KTP, without deterioration or progression of CD. In this case, CD did not prevent KTP; therefore, more active KTPs should be recommended for CD patients based on this finding.



 XML Download
XML Download