

 PDF
PDF ePub
ePub Citation
Citation Print
Print


The occurrence of pheochromocytoma might be sporadic or might have a genetic basis. Genetic pheochromocytoma is associated with multiple endocrine neoplasia (MEN) type 2A, MEN type 2B, and von Hippel-Lindau (VHL) disease. MEN 2 includes autosomal, dominantly inherited disorders manifesting as medullary thyroid cancer (MTC) and pheochromocytoma with primary hyperparathyroidism (MEN 2A) or mucosal neuromas (MEN 2B). VHL disease is an autosomal dominant disorder caused by heterozygous mutations in the VHL tumor suppressor gene. VHL disease is characterized by hemangioblastomas of the retina; hemangiomas of the adrenal gland, liver, and lung; and a variety of solid tumors, including clear-cell renal carcinomas, pheochromocytomas, paragangliomas, and pancreatic neuroendocrine tumors.1 RET mutations are identified in more than 90% of index cases of MEN2 and familial medullary thyroid cancer (FMTC) and in about 4–12% of apparent sporadic cases.2 Here, we report a 54-year-old man presenting with pheochromocytoma and renal cell carcinoma, who was carrying a novel missense RET mutation. We also provide a relevant literature review on the topic.
CASE
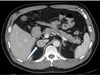
A 54-year-old man visited our endocrine department to investigate a right adrenal mass incidentally detected on abdominal ultrasonography performed for routine health screening. Abdominal computed tomography (CT) revealed a 3-cm solid and homogenous mass in the right adrenal gland (Fig. 1). He had a medical history of cavernous angioma. Brain MRI revealed cavernous sinus malformation in the right upper medial pons. He had been treated for essential hypertension and gout for the last 2 years. His blood pressure was 130/80 mmHg, and he did not complain of abdominal symptoms.
Laboratory evaluation yielded the following results. The serum dopamine, norepinephrine, and epinephrine levels were 0.022 ng/mL (reference range, 0–0.2 ng/mL), 0.226 ng/mL (reference range, 0–0.8 ng/mL), and 0.037 ng/mL (reference range, 0–0.3 ng/mL), respectively. His 24-h urine vanillylmandelic acid (VMA) and meta-nephrine levels were 8.8 mg/day (reference range, 0–8.0 mg/day) and 3.010 mg/day (reference range, 0–1.3 mg/day), respectively, and 24-h urine-free cortisol was 112.8 Og/day (reference range, 55.5–286.0 Og/day). Serum cortisol and plasma ACTH levels were 0.967 Og/dL (normal, 2.3~19.4 Og/dL) and 1.57 pg/mL (normal, 5~60 pg/mL), respectively, after overnight dexamethasone suppression.
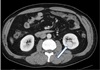
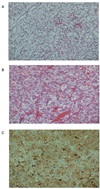
A whole-body scan with iodine-131-meta-iodobenzylguanidine (I-131 MIBG) was performed, which revealed focal tracer uptake in the right adrenal area and normal tracer uptake in other organs. During evaluations, we could also find a 2.5-cm enhancing mass on the lower pole of the left kidney (Fig. 2) on the same abdominal CT, and the mass was suspected to be renal cell carcinoma. Laparoscopic right adrenalectomy and left radical nephrectomy were performed. The left renal mass was diagnosed as renal cell carcinoma (Fig. 3A) and the right adrenal mass as pheochromocytoma (Fig. 3B) by histopathologic evaluation.
We performed genetic testing using the patient's blood to investigate mutations in RET and VHL genes. VHL tumor suppressor gene mutation analysis revealed no mutation; however, RET gene mutation analysis identified a novel missense mutation (exon 15, 2611G>A, Val871IIe) (Fig. 4). RET immunostaining was negative for S-100 in renal cell carcinoma tissue, but pheochromocytoma tissue was positive (Fig. 3C).
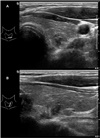
According to this result, we performed neck CT, which showed mild enlargement of both parathyroid glands. The serum calcium level was 10 mg/dL (reference range, 8.2–10.2 mg/dL), and parathyroid hormone level was 31 pg/mL (reference range, 10–65 pg/mL). Thyroid ultrasonography revealed a 0.6-cm hypoechoic nodule in the left thyroid gland (Fig. 5). Serum calcitonin was < 2 pg/mL (reference range, < 8.4 pg/mL). Ultrasound-guided fine-needle aspiration (USG-FNA) from the left thyroid nodule showed no malignant cells. However, the calcitonin level of the wash-out fluid of the nodule was 51.3 pg/mL.
Although there are no prophylactic thyroidectomy guidelines regarding this novel missense RET mutation, we recommended total thyroidectomy for the left thyroid nodule. In the pathologic specimen, papillary thyroid cancer was detected incidentally in the right thyroid gland, but no medullary thyroid cancer or hyperplasia was detected in the left thyroid gland.
DISCUSSION
The clinical practice guidelines published by the American Endocrine Society in 2014 recommends that all patients with pheochromocytoma and paragangliomas (PPGLs) should be engaged in shared decision making for genetic testing. Since 1990, 14 different PPGL susceptibility genes have been reported, namely, NF1, RET, VHL, SDHD, SDHC, SDHB, EGLN1/PHD2, KIF1β, SDH5/SDHAF2, IDH1, TMEM127, SDHA, MAX, and HIF2α. There are several reasons that justify genetic testing in patients presenting with PPGLs. First, at least one-third of all patients with PPGLs have disease-causing germline mutations. Second, SDHB mutations lead to metastatic disease in 40% or more of affected patients. Third, establishing a hereditary syndrome in the proband may result in earlier diagnosis and treatment of PPGLs and other syndromic manifestations in relatives.3 In clinical practice, patients with PPGLs can present with features that indicate a high likelihood of a hereditary cause. Such features include a positive family history (based on family pedigree or identification of a PPGL-susceptibility gene mutation in a relative); syndromic features; and multifocal, bilateral, or metastatic disease.4
The present patient showed multifocal and syndromic features. In addition to the presence of pheochromocytoma, the patient was diagnosed with cerebral cavernous hemangioma and renal cell carcinoma. These combinations of multiple disorders and syndromic features made us consider genetic testing. Although RET mutation is primarily well-known to be associated with the development of hereditary MTCs, including MEN2 and FMTC, it is also known to stimulate the development of pheochromocytoma and PPGLs by the overexpression of genes underlying embryonic development of the enteric nervous system and the kidney by encoding ligand-independent activation of tyrosine kinase,56 Therefore, we first performed genetic testing to investigate RET mutations, which revealed a novel missense RET mutation (exon 15, 2611G>A, Val871IIe).
VHL syndrome is an autosomal dominant disorder caused by deletions or mutations in a tumor suppressor gene mapped to human chromosome 3p25. Clear-cell renal cell carcinoma occurs in up to 70% of patients with VHL, and pheochromocytomas occur in association with specific alleles of the VHL gene.7 Early detection and treatment of VHL syndrome are important, because if left untreated, it may result in blindness or permanent brain damage. Most of these patients die from complications of renal cancer and pheochromocytoma.89 On the basis of multiple features compatible with VHL syndrome, we also tested for mutations in VHL, but the result was negative. RET immunostaining showed negative staining in renal cell carcinoma tissue. This implies that RET mutation is not a cause of renal cell carcinoma.
After right adrenalectomy and left radical nephrectomy, we performed thyroid ultrasonography. The result of USG-FNA showed no malignant cells except scanty follicular cells in the left thyroid nodule. Elevated calcitonin level in the wash-out fluid of the nodule (51.3 pg/mL). reflected a high sensitivity of MTC; therefore, we could not exclude the possibility of MTC.10
Several studies have suggested prophylactic thyroidectomy in patients with positive RET gene mutation. In a single-center study, 75 patients with preoperatively proven positive RET mutation underwent prophylactic thyroidectomy, and histopathology results revealed MTC in 46 patients (61%).11 The American Thyroid Association (ATA) 2009 guidelines classify RET protooncogene mutation carriers into 4 levels depending on gene analysis, and risk factors of MTC and recommendation of prophylactic thyroidectomy are also graded into 4 levels each.12 However, no absolute recommendation on prophylactic thyroidectomy for a case like ours is available in the literature, because it involves a novel RET mutation. Although our patient had no family history of MTC and no evidence of malignancy from FNA, prophylactic thyroidectomy was performed based on a RET gene mutation and high calcitonin levels from wash-out fluid.
To conclude, we identified a new RET mutation in a patient presenting with pheochromocytoma and incidentally detected renal cell cancer. We suggest that sporadic pheochromocytoma with other malignant tumors might necessitate further evaluation by gene mutation screening.

 XML Download
XML Download