

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Autosomal-dominant mutations in the prion protein gene, ‘PRNP’, lead to genetic prion diseases that account for 10–15% of human prion diseases.12 Due to differences in clinical and neuropathological features, human genetic prion diseases are categorized into three groups: genetic Creutzfeldt-Jakob disease (CJD), Gerstmann-Sträussler-Scheinker disease (GSS), and fatal familial insomnia.1
The history of GSS dates back to 1936, when an Austrian family with GSS was reported by J. Gerstmann, E. Sträussler, and I. Scheinker.34 In 1995, a proline-to-leucine mutation at codon 102 (P102L) in the PRNP gene was identified in this family.4 More than 16 different point mutations (including P84S, P102L, P105L, P105S, A117V, G131V, S132I, V176G, H187R, F198S, D202N, E211D, Q212P, Q217R, Y218N, and M232T) and several octapeptide repeat insertions in PRNP are now linked to GSS, with P102L reportedly to be the most common worldwide.5678910 P102L-associated GSS (henceforth P102L GSS) was also the most commonly identified GSS in a Chinese population.11
The most-common phenotype of GSS patients is slowly progressing cerebella ataxia initially, with parkinsonism and dementia appearing later in the clinical course.1213 The presence of multicentric prion protein amyloid plaques in neuropathology remains the key feature of GSS that differentiates it from most of the other genetic prion diseases.314 The survival times of GSS patients reportedly vary from 1 to 10 years worldwide.3 However, some patients with symptoms more typical of sporadic CJD (sCJD) have a much shorter clinical course.151617
There have been 12 P102L GSS cases diagnosed by the Chinese National Surveillance Network for CJD under the framework of the Chinese Center for Disease Control and Prevention (CCDC) since 2006, constituting about 8% of all diagnosed genetic prion diseases. Here we investigated the epidemiological, clinical, genetic, and laboratory characteristics of these P102L GSS patients.
METHODS
Clinical data collection and case identification
Twelve Chinese P102L GSS cases were enrolled in this study. Information was obtained from the Chinese National Surveillance Network for CJD under the leadership of the CCDC as described previously.18 Briefly, the appearance of periodic sharp wave complexes (PSWC) was regarded as a prion-disease-specific EEG abnormality. MRI findings were considered to be abnormal in the presence of high signal intensities in the caudate/putamen and/or symmetrical or dissymmetrical cortical ribbon syndrome in diffusion-weighted imaging (DWI). The geographical distribution was determined based on the registered permanent addresses of the patients. The survival time was calculated as the duration from disease onset to death (analyzed from January 1, 2005 to April 2, 2018). The final diagnosis was made according to the diagnostic criteria for CJD issued by the China National Health Commission.19
Laboratory tests
Blood and CSF were collected by physicians in local hospitals and transferred to the center. Western blotting of CSF for 14-3-3 protein and PRNP sequencing were conducted following the standard operating procedure of the Chinese National Surveillance Network for CJD.1820 In brief, a 20-µL CSF sample was separated in 12% sodium dodecyl-sulfate polyacrylamide gel electrophoresis and immunoblotted with a 14-3-3-specific monoclonal antibody (1:500 diluted; SC-133233, Santa Cruz; https://www.scbt.com/scbt/product/pan-14-3-3-antibody-b-8). The genomic DNA of the patients was extracted from peripheral blood samples for sequencing analysis of PRNP and the polymorphisms of codons 129 and 219. All experiments in which mutations were identified by comparisons with the standard (NCBI: NM-183079.1) were repeated at least once using new blood samples in order to avoid misreading.
Statistical analyses
The statistical analyses were performed using SPSS 20.0 statistical software (IBM Corp., Armonk, NY, USA). Categorical variables were compared using Fisher's exact test, while the relationship between disease duration and the speed of dementia progression was analyzed using logistic regression. Probability (p) values less than 0.05 were considered to be statistically significant. Survival analyses were performed using GraphPad Prism software (GraphPad Software, San Diego, CA, USA).
Ethics statement
The study was approved by the Research Ethics Committee of National Institute for Viral Disease Control and Prevention, CCDC (approval no. #2013031). Written informed content was obtained from a family member or relative of each patient in accordance with the requirements of the Chinese National Surveillance Network for CJD.
RESULTS
General information
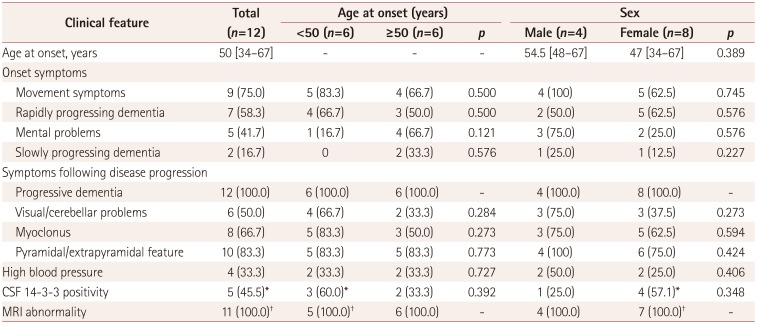
According to the data of the Chinese National Surveillance Network for CJD, which started officially in 2006, 12 P102L GSS patients were identified and diagnosed by April 2018, all of whom were Han Chinese. Based on their permanent addresses, most of the patients came from the eastern part of China: three from Shandong province, and one from each of Shanxi, Henan, Hebei, Jiangxi, Anhui, Hubei, Jiangsu, Zhejiang, and Jilin. Clinical interviews did not reveal any blood relationship among these patients. There were four males and eight females, giving a sex ratio of 1:2. The median onset age was 50 years, ranging from 34 to 67 years. Most of the patients were aged 40–49 years (n=5), followed by 50–59 years (n=3) and 60–69 years (n=3) (Fig. 1).
Clinical features
Most of the patients presented with more than one symptom at the onset, which included movement symptoms (gait and walking instability), mental problems (e.g., anxiety, dystrophy, irritability, and emotional lability), rapid or slow progression of memory decline, and dementia. As indicated in Table 1, movement symptoms were reported in 75% of the cases (n=9). The proportion of movement problems was slightly higher for an onset age of <50 years (5/6, 83.3%) than for one of ≥50 years (4/6, 66.7%). Rapidly progressing dementia was reported in 7 cases (58.3%). Mental problems such as anxiety and irritability appeared in 5 cases (41.7%), and were more common for an onset age of ≥50 years (4/6, 66.7%) than for one of <50 years (1/6, 16.7%). Additionally, there were 2 cases of slowly progressing dementia, and 4 cases with a medical history of high blood pressure lasting for 1–4 years based on the normal values of blood pressure for various ages of Chinese subjects.
Along with the disease progression, more sCJD-associated neurological symptoms and signs appeared. Progressive dementia was recorded in all cases (12/12, 100%). Pyramidal and extrapyramidal symptoms (10/12, 83.3%), myoclonus (8/12, 66.7%), and visual or cerebellar problems (6/12, 50%) also appeared frequently. Akinetic mutism was observed in the late stage of disease in 11 cases (Supplementary Table 1 in the online-only Data Supplement). No statistical differences were found in the comparisons between the symptoms and the groups of gender or onset age.
Clinical examination and laboratory tests
EEG was applied to 8 cases at least once during hospitalization, two of which exhibited PSWC. Eleven of the 12 cases received MRI scanning, which revealed sCJD-specific abnormalities in all cases. Three cases had high signal intensities in the caudate/putamen, 3 cases had DWI ribbon-like signals, and 5 cases had both abnormalities (Supplementary Table 1 in the online-only Data Supplement). There were no statistically significant differences in the distributions of specific EEG and MRI abnormalities between the groups of gender and onset age. Lumbar puncture was performed in 11 patients, and the routine biochemistry values were all within the normal ranges. Western blotting for 14-3-3 protein revealed positive bands in the CSF samples from 5 cases (45.5%) (Table 1).
Survival time
Up to April 2018, 7 cases had died on a known date, 4 were still alive, and one was lost to follow-up. The survival times based on different sex and onset ages are shown in Fig. 2. The clinical course of the 7 dead cases varied from 10 to 44 months, with a median of 16 months. Five of the 7 cases (71.4%) with a short survival time (less than 2 years) complained of rapidly progressing dementia at the onset, while the other two patients with a long duration (longer than 40 months) displayed relatively slowly progressing dementia. Moreover, female and younger patients at onset seemed to show shorter survival times than males and older-onset patients (Fig. 2). However, no statistical significance was found, which was possibly due to the small number of cases.
PRNP sequencing and family history
PRNP sequencing confirmed that all 12 cases were carrying the P102L mutation in 1 PRNP allele. All cases had the methionine homozygous genotype at codon 129 (M129M), while 10 cases were glutamic acid homozygous at codon 219 (E219E) and one was glutamic acid/lysine heterozygous at codon 219 (E219K) (Supplementary Table 1 in the online-only Data Supplement). No other mutation was noted in the remaining PRNP sequences analyzed.
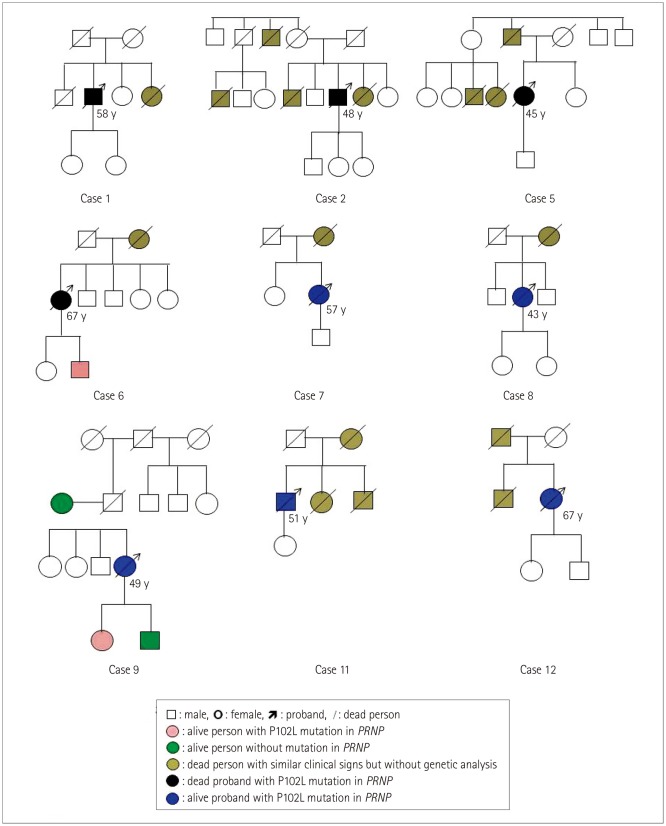
Carefully tracing of the disease-related family histories identified that the parent and/or sibling of 10 cases had died of similar neurological problems without a definite diagnosis. Only 2 cases (Cases 3 and 10) denied having a relevant family history. As shown in Fig. 3, Cases 6, 7, and 8 recalled similar symptoms only in the generation of their parents, while in Case 1 this was only in the proband generation. Cases 2, 5, 11, and 12 had other close family members who had died with neurological symptoms both in their parent and proband generations. Cases 3 and 4 reported a disease-related family history during hospitalization, but their family members refused to provide detailed information. Only four family members from two families donated blood samples for PRNP sequencing: three from Case 9 who denied having a disease-related family history, and one from Case 6. PRNP sequencing verified that the mother and son of Case 9 had a wild-type PRNP genotype, while her daughter was a P102L missense mutation carrier. The son of Case 6 also carried the P102L mutation. Both of these two P102L mutation carriers were M129M and E219E homozygous, and had not exhibited any neurological abnormality up to April 2018.
DISCUSSION
This study systematically evaluated the epidemiology, clinical, genetic, and laboratory features of 12 Chinese P102L GSS patients. There were more female than male patients, but neither the clinical nor laboratory examination revealed any sex-associated difference. Like the P102L GSS cases reported worldwide,23 the onset ages for the present Chinese patients were relatively young, with a median of 50 years. However, the average disease duration of the seven Chinese patients seems to be shorter than the global value (52 months, ranging from 7 to 132 months).101621 Patients showing the sCJD phenotype (rapidly progressing dementia) seem to have a shorter survival time (10–16 months). It is particularly notable that patients with a prominent psychiatric onset usually have an early onset and death.2 The youngest Chinese P102L GSS patient in the present study, a 34-year-old woman with a psychiatric family history, also had a short survival time (11 months) and had mental syndrome as her initial symptoms. Whether and why Chinese GSS patients have relatively short survival needs to be investigated in more cases.
With regard to other symptoms, cognitive decline is generally mild in P102L GSS cases and this aggravates during disease progression.21 Three-quarters of the present Chinese patients had early cognitive deficits, and the progression of dementia was relatively rapid in most of them, showing an sCJD-like phenotype. Among 57 Japanese P102L GSS cases, 21% presented with an sCJD-like phenotype with early and prominent dementia.17 Another study found that 40% of cases showed cognitive symptoms at the onset.10 Whether the higher proportion of cognitive problems is a characteristic of Chinese P102 GSS patients needs to be investigated further in more cases.
The sCJD-associated abnormalities in MRI appear frequently in Chinese cases of P102L GSS, especially in the relatively late stage. High signal intensities in the caudate/putamen and DWI ribbon-like signals are equally observable. A high proportion (80%) of MRI abnormalities was found in Japanese GSS patients with a rapidly progressing clinical phenotype.17 However, a investigation based on data obtained in the EuroCJD study found fluid-attenuated inversion recovery or DWI hyperintensities in the basal ganglia in 30% of the P102L GSS cases.16 Other studies have even noted cortical ribboning or deep nuclei hyperintensities to be very uncommon.22 The significance of MRI findings in P102L GSS therefore needs further evaluation. Only one-quarter and less than half of the Chinese patients in the present study were verified as having PSWC in EEG and positivity for 14-3-3 protein in the CSF, respectively. Coincidental, PSWC in EEG and 14-3-3 positivity in the CSF were observed in 50% and 31% of Caucasian GSS patients, respectively.23 It therefore appears that diagnosing P102L GSS based on EEG and CSF 14-3-3 positivity will result in fewer cases than when applying MRI.
The disease-associated family history of P102L GSS patients seems to vary greatly. The EuroCJD study involved 52 cases in European countries and found a family history in 69.7% of them.23 However, other studies involving German and Japanese patients found positive family histories in 84% to 100% of cases.1617 A high positivity rate (83.3%) for the family history was found in the present Chinese P102L GSS patients. It should be noted that the disease-related family histories for the patients in this study were obtained not only from routine clinical interviews performed during hospitalization but also during the subsequent surveillance follow-up. Therefore, careful and repeated interviews with patients and family members are highly recommended.
Polymorphism of PRNP is associated with both the susceptibility and phenotype of prion diseases.2425 The polymorphism at PRNP codon 219 with the substitution of glutamate for lysine seems to be found only in Asian population, and not in Caucasians.2627 P102L GSS patients with this polymorphism have been described as showing different clinical and pathological features.27282930 The lysine at codon 219 is believed to be protective against sCJD. Only one of the present P102L GSS case was E219K heterozygous; however, he displayed a sCJD phenotype with rapidly progressing dementia, PSWC in EEG, and a survival time of 10 months after the onset. Furthermore, this patient seemed to show less clinical symptoms than the other cases. More evidence is needed for exactly how the polymorphism at codon 219 influences on the clinical and pathological characteristics of P102L GSS in Chinese subjects.
In summary, based on data collected by the Chinese National Surveillance Network for human prion diseases from 2006 to 2018, P102L is the most-common GSS-associated mutation. More than half of the identified patients had the sCJD phenotype. Due to the complexity of clinical manifestations of GSS patients, PRNP sequencing is an indispensable tool for diagnosing GSS.


 XML Download
XML Download