

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Most cases of acute leukemia (AL) are classified as either myeloid or lymphoid lineage based on the expression of a set of antigens. However, there are cases of patients with blasts expressing both lymphoid and myeloid features at diagnosis, known as biphenotypic acute leukemia (BAL) or mixed-phenotype acute leukemia (MPAL) [1]. These divergent morphologic and immunophenotypic features may be uniformly present in one blast population (biphenotypic leukemia) or may be seen on distinct blast populations in a single patient (bilineal leukemia) [2].
The European Group for Immunological Classification of Leukemias (EGIL) proposed a set of diagnostic criteria for BAL. This scoring system was based on the number and degree of specificity of certain makers for myeloid or T/B lymphoid blasts [34]. In 2008, the new World Health Organization (WHO) criteria were proposed for the classification of acute leukemia of ambiguous lineage (ALAL), which include significant modifications of the diagnostic criteria for AL of mixed phenotype. Cases with no lineage-specific markers were designated as acute undifferentiated leukemia (AUL), which often express CD34, HLA-DR, and/or CD38, and sometimes TdT, but lack specific myeloid or lymphoid antigens [56]. A type of leukemia with blasts that co-expresses certain antigens of more than one lineage on the same cells or that has separate populations of blasts of different lineages is referred to as MPAL. They can be further designated as B-myeloid or T-myeloid based on flowcytometric immunophenotyping, irrespective of whether one or more than one population of blasts was found. Thus, MPAL includes both biphenotypic and bilineal ALs. These WHO criteria were more stringent than those of the EGIL criteria and relied heavily on positivity for myeloperoxidase (MPO) [5]. In 2016, the WHO classification was updated but the criteria for ALAL remained unchanged [6].
The exact incidence of BAL or ALAL in childhood is unknown. The optimal therapy for this subtype of leukemia has not been established, especially in pediatric patients. Little is known about the prognostic implications of immunophenotypes of BAL [7]. The role of hematopoietic stem cell transplantation (HSCT) in first remission remains contentious [28910].
The present study aimed to retrospectively investigate the incidence and clinicopathological characteristics of pediatric BAL or ALAL patients in Gwangju and Chonnam province from 2001 to 2017.
MATERIALS AND METHODS
Patients
From January 2001 to December 2017, the medical records of children newly diagnosed of BAL based on the EGIL criteria or ALAL based on the 2008/2016 WHO criteria who were admitted at the Chonnam National University Hospital (CNUH) and Chonnam National University Hwasun Hospital (CNUHH) were retrospectively reviewed. Patients' baseline clinicopathological data such as age, sex, blood counts, blast % in peripheral blood (PB), bone marrow (BM) morphology, immunophenotyping, and cytogenetic/molecular studies were obtained (Tables 1, 2). Treatment methods and outcome data regarding the induction chemotherapy regimen, response to chemotherapy, use of HSCT, relapse, and death were reviewed. This study was an observational, retrospective, descriptive study of the clinical aspects of childhood BAL or ALAL, which was approved by the Institutional Review Board of the CNUHH (CNUHH-2018-148).
Immunophenotyping
Immunophenotyping of BM aspirates was performed using the consensus method. The result was considered positive if the antigen was expressed on more than 20% (10% for cytoplasmic markers, anti-MPO, and TdT) of leukemic cells. The panels of monoclonal antibodies used in flow cytometric immunophenotyping to detect B-cell, T-cell, and myeloid antigens as follows: myeloid lineage (anti-MPO, CD13, CD14, CD33, CD64, and CD117), megakaryocytes (CD41 and CD61), natural killer cells (CD56), lymphoid lineage (CD10 and TdT), T-lymphoid lineage (CD2, surface CD3, cytoplasmic CD3, CD4, CD8, CD5, and CD7), B-lymphoid lineage (CD19, CD20, and cytoplasmic CD22), and hematopoietic precursor (CD34 and HLA-DR).
Diagnostic criteria
The diagnostic workup of BAL was based on initial morphological, cytochemical, and immunophenotypic evaluation of the BM. Using the EGIL scoring system, BAL diagnosis was established when the score from two separate lineages was greater than 2 [34].
The requirements for assigning specific lineages to the blasts were given in the 2008/2016 WHO criteria. Only a limited number of antigens were used in defining the pattern of lineage involvement. The myeloid lineage defining marker was MPO as detected by flow cytometry, immunohistochemistry, or cytochemistry; and monocytic differentiation was required for assigning diffuse positivity for non-specific esterase or expression of at least two of the following: CD11c, CD14, CD36, CD64, and lysozyme. The T-lineage defining markers were cytoplasmic CD3 or surface CD3. The B-lineage defining marker was either a strong CD19 with at least one of the strongly expressed CD79a (cytoplasmic CD22, CD10, or weak CD19) or a weak CD19 with at least two of the strongly expressed CD79a (cytoplasmic CD22 and CD10). AUL include leukemias that express no lineage specific markers. In addition, the 2008/2016 WHO classification includes certain types of MPAL harboring Philadelphia chromosome (Ph1) or MLL rearrangements as distinct diagnostic subgroups [5111213].
Treatment protocols
Patients were initially treated with remission induction therapy of either acute lymphoblastic leukemia (ALL)- or acute myelogenous leukemia (AML)-directed chemotherapy. ALL-directed regimen was used in six patients. The Children Cancer Study Group 1882/1901 regimen based on vincristine (VCR), prednisone (PRD), and L-asparaginase (L-asp) with or without daunorubicin was used in three patients, while the Korean High Risk (HR) ALL regimen including a four-drug induction therapy was used in two since 2012. Interfant-99 hybrid regimen consisting of VCR, PRD, L-asp, and cytarabine was used in the patient with congenital leukemia (UPN #12). For AML-directed regimen, the KSBRM induction regimen consisting of idarubicin (IDA) plus N4-behenoyl-1-β-D-arabinofuranosyl cytosine was given in three patients, while the Korean AML 2012 regimen, a double induction regimen consisting of IDA or mitoxantrone plus cytarabine based chemotherapy, was used in three.
Statistical analysis
Continuous variables were expressed as means±standard deviations; categorical variables were expressed as numbers and percentages. Continuous variables were compared using the Student's t-test, while categorical variables were compared using the chi-square test or Fisher's exact test. Probabilities of survival were estimated using the Kaplan-Meier (K-M) method and were compared using log-rank test. Prognostic variables were evaluated by multivariate analyses using a Cox regression proportional hazard model. A P-value of <0.05 was considered significant. The software package SPSS version 21.0 (SPSS Inc, Chicago, IL, USA) was used for all statistical analyses.
RESULTS
Patients
Among 377 pediatric patients diagnosed with AL, 13 satisfi ed the definition of BAL based on the EGIL criteria or ALAL based on the WHO criteria. However, a patient with secondary malignancy following chemotherapy for non-Hodgkin's lymphoma 9 years earlier was excluded; thus, only 12 primary cases (3.2%) were finally enrolled in this study (Fig. 1). Eleven patients were diagnosed with BAL based on the EGIL criteria. The patient with UPN #12 was excluded as it was considered an undefined case. With the more stringent 2008/2016 WHO criteria, 7 (1.9%) patients remained as ALAL (AUL, 2; MPAL). Six BAL patients were redirected as ALL (N=3; UPN #1, #7, #10), AML (N=2; UPN #2, #5), and AUL (UPN #9) based on the WHO criteria (Table 2, Fig. 1).
Of 11 patients with BAL, 6 (54.5%) had a B-lymphoid/myeloid phenotype, while 5 (45.5%) had a T-lymphoid/myeloid phenotype based on the EGIL criteria (Table 2). Only 5 (1.3% of the total cohort) of these patients were categorized as having MPAL according to the 2016 WHO criteria (B-lymphoid/myeloid, 3; T-lymphoid/myeloid, 2). UPN #4 with B-lymphoid/myeloid phenotype was reclassified as MPAL with t(9;22) by the WHO. Likewise, UPN #7 with T-lymphoid/myeloid was classified as MPAL with MLL rearrangement. Additionally, two patients were classified as AUL based on the WHO criteria, and one of them had MLL rearrangement (UPN #12) (Table 2).
The characteristics of patients are presented in Table 1. The median age of the patients at diagnosis was 8.6 years (range, 0 mo to 17.8 yr). Of the 11 patients, 7 (58.3%) were men and 5 (41.7%) were women. The median initial WBC count was 119.5×109/L (range, 1.8–800.9×109/L), and four patients had WBC count higher than 100×109/L. The median PB blast at diagnosis was 41.9% (range, 0–96.0%), and the median BM blast at diagnosis was 72.3% (range, 26.0–98.0%). None of the patients had CNS disease at presentation. The clinical characteristics of BAL patients with either B-lymphoid/myeloid or with T-lymphoid/myeloid phenotypes are described in Table 3.
Immunophenotypic characteristics
Table 2 shows the immunophenotypic characteristics of each BAL or ALAL patient. The expression of immunological and cytochemical markers on leukemic blasts according to BAL subtypes is summarized in Table 4. In 12 patients with myeloid lineage disease, CD13 was positive in 8 (66.7%), CD33 in 7 (58.3%), and anti-MPO in 5 (41.7%). The B-lymphoid lineage markers were cytoplasmic CD22 (5/12; 41.7%), CD19 (4/12; 33.3%), and TdT (4/12; 33.3%). The most frequently associated positive T-lymphoid lineage marker was CD7, which was positive in all four patients (100%). Interestingly, CD2 and CD7, T-lymphoid markers, have been aberrantly observed in B-lymphoid/myeloid cases (3/6, 50.0%). The stem cell markers, such as HLA-DR and CD34, were positive in 10 (83.3%) of 12 patients.
Cytogenetic characteristics
Results of cytogenetic analysis performed on all patients were available. Details of cytogenetic analysis and molecular studies are presented in Table 1. In cytogenetic analysis, 3 (25.0%) patients had normal karyotypes, while the remainder (75.0%) showed a clonal abnormality. Abnormalities involving the MLL gene at the 11q23 locus were found in two patients (UPN #7 and #12). The rest had t(9;22) (q34;q11.2) (Ph1+) and BCR-ABL+ (UPN #4).
Treatment outcome
Six patients initially received standard induction therapy for AML (KSBRM induction, 3; AML 2012 1st induction, 3), whereas six received ALL-directed induction therapy (CCG 1882/1901 induction, 3; Korean HR ALL induction, 2; Interfant-99 induction, 1) (Table 5). Overall, 7 (58.3%) of 12 patients achieved complete remission (CR) after their initial induction therapy. In the AML induction group, only 1 (16.7%) achieved a CR, while 2 (33.3%) and 3 (50.0%) had partial remission and non-remission, respectively. By contrast, all patients (6/6, 100.0%) achieved a CR after ALL-directed chemotherapy. Thus, ALL-directed induction chemotherapy was associated with higher chances of achieving a CR compared with AML-directed regimens (100.0% vs. 16.7%; P=0.015). According to BAL phenotype, the CR rate was similar in M+T (3/5, 60.0%) vs. in M+B (3/6, 50.0%; P=1.0).
Moreover, three of five patients who failed to respond to initial AML therapy attained a CR after receiving salvage standard ALL induction therapy (UPN #3, #9, and #11). The remaining two who failed to respond to AML therapy attained a CR after receiving FLAG-IDA (UPN #1 and #2). Thus, all 12 patients eventually achieved a CR either after initial induction or salvage induction chemotherapy.
Table 5 shows the remission induction regimens, salvage regimens, post-remission therapy, and survival outcomes of patients. Interestingly, among six patients whose diagnosis were subsequently changed from BAL, two of three patients who were initially treated with ALL-directed induction therapy survived (66%; UPN #7, #10) while three who had AML-directed therapy succumbed (0%; UPN #1, #2, and #9). Nine (75%) patients eventually underwent an allogeneic HSCT [three in the first CR (CR1) after initial induction regimen (UPN #6, #8, and #10), three in CR1 after salvage chemotherapy (UPN #3, #9, and #11), and three in the second CR (CR2) after relapse (UPN #1, #2, and #4)] (Fig. 1). Three patients did not undergo HSCT. Patients with UPN #7 who received chemotherapy alone had continuous CR for over 7 years. By contrast, one patient died of intracerebral hemorrhage (UPN #5), while the other patient had disease progression (UPN #12).
Survival rates
At a median follow-up of 5 years (range, 0 mo–10 yr), the OS was 51.1±15.8% (Fig. 2A), and the EFS was 51.9±15.7% in 11 patients with BAL based on the EGIL criteria (Fig. 2B). Fig. 3 demonstrates the survival comparison between AUL and MPAL based on the WHO criteria. All AUL patients (N=2) died at 5 and 8 months after diagnosis. The 5-year OS and EFS were 0.0% in AUL and 75.0±21.7% in MPAL (P=0.008) (Fig. 3A, B).
Univariate analyses of clinicolaboratory variables for the prediction of prognosis were performed on initial leukocyte count, immunophenotype (M+B vs. M+T), cytogenetic abnormality, type of initial induction therapy (ALL directed vs. AML directed), and the use of HSCT. The initial leukocyte count <100×109/L was associated with better 5-year OS or 5-year EFS (both for 62.5±17.1% vs. 0.0%, P=0.278, Fig. 4A and B, respectively). T-lymphoid/myeloid BAL seemed to have a better OS and EFS than B-lymphoid/myeloid cases (both for 80.0±17.9% vs. 33.3±19.2%, P=0.204, Fig. 5A and B, respectively).
The survival rates were compared according to the type of initial induction therapy (Fig. 6). The 5-year OS probability for ALL-directed induction therapy was 50.0±20.4%, while that for AML-directed induction therapy was 41.7±22.2% (P=0.874; Fig. 6A). The 5-year EFS for ALL-directed induction therapy was 50.0±20.4%, while that for AML-directed induction therapy was 44.4±22.2% (P=0.995; Fig. 6B).
The survival of all 12 patients with BAL or ALAL who underwent HSCT is shown in Fig. 7. The 5-year OS probability of patients who underwent HSCT was 50.8±17.7%, while that of patients who received chemotherapy alone was 33.3±27.2% (P=0.316; Fig. 7A). Furthermore, the 5-year EFS of patients who underwent HSCT was 51.9±17.6%, while that of patients who underwent chemotherapy alone was 33.3±27.2%, (P=0.224; Fig. 7B).
DISCUSSION
The incidence of BAL or ALAL varies according based on the criteria (EGIL or 2016 WHO) used for the diagnosis. BAL is reported to account for 2–5% of all ALs based on the EGIL criteria, while ALAL 1–2.5% based on the WHO criteria [2914], which are similar to the current study of 2.9% and 1.9%, respectively. Additionally, the incidence of AUL and MPAL among ALAL was 0.5% and 1.3%, respectively, in this study. Although the incidence was not clearly defined, the incidence of AUL was reported to be 0.2–1% in some studies [1516].
In contrast to the EGIL approach of scoring a detailed blast immunophenotype with numerous markers, the WHO criteria emphasize a few key lineage-defining markers with particular emphasis on CD19 for B lineage, CD3 for T lineage, and MPO for myeloid lineage. The WHO approach is simpler but relies heavily on the sensitivity and specificity of a few markers. Moreover, the WHO classification does not specify thresholds for positivity of these key markers, leaving it up to individual laboratories to decide on the definition of significant expression [13]. In addition, the WHO diagnostic criteria defined AUL as a type of leukemia that does not involve the expression of those specific antigens and hence set up the classification of ALAL, including MPAL and AUL. In the present study, the incidence might have been underestimated as some immunological markers such as cyCD79a were not tested in our institute.
The phenotype distribution in our patients was no different from those reported in previously published studies. The number of patients having the B-lymphoid/myeloid phenotype was more than the number of patients having the T-lymphoid/myeloid phenotype [217]. In adult studies, more than 70% of BAL patients had the B-lymphoid/myeloid phenotype, whereas only 23–33% had the T-lymphoid/myeloid phenotype. The B-lymphoid/T-lymphoid/myeloid phenotype and B-lymphoid/T-lymphoid phenotype were extremely rare [7181920].
In the WHO 2016 diagnostic criteria, t(9;22)/Ph+ positive and MLL rearrangement were classified separately. Although MPAL was not associated with a consistent cytogenetic/molecular abnormality, the Philadelphia chromosome was the most common abnormality, with 17–41% incidence in BAL and MPAL patients with almost exclusively B-lymphoid/myeloid phenotype [71421]. BCR-ABL1 was more common among adult MPAL patients (15%) than among pediatric MPAL patients (3%) [141522]. However, all Ph+ patients should be identified as quickly as possible because they may benefit from the addition of tyrosine kinase inhibitors (TKI) same as those with Ph+ ALL [122324].
The second most common cytogenetic abnormality was MLL rearrangement as reported in other studies. Its incidence ranged from 10% to 15% [7814] and were higher in the pediatric age group than in the adult group (15% vs. 3%) [141525]. In the present study, the incidence of cytogenetic abnormality was also 16.7%. No single chromosomal abnormality is unique to ALAL. However, the present data and those of other studies showed that structural abnormalities are common.
MPALs may have worse prognosis than other types of leukemia. The proposed reasons were as follows: the mixed-phenotype leukemic stem cells are chemoresistant owing to slow replication, the blasts can adapt to therapy by switching phenotype, and some MPALs express high levels of multidrug resistance proteins [1226].
Because selection of an anti-leukemic chemotherapy regimen for AL is largely based on whether a case is classified as myeloid or lymphoid, the presence of markers for both lineages may have important implications for treatment. Thus, there are no agreed chemotherapy protocols for patients with ALAL as yet [1227]. In the current study, the induction regimen was selected based on the morphology of the blasts and cytochemical stains. All 6 (100.0%) patients who underwent ALL induction therapy reached a CR. On the contrary, among six patients who received AML induction therapy along, 1 (16.7%) achieved a CR after one cycle, while 2 achieved a CR after FLAG-IDA salvage chemotherapy. The remaining three patients reached a CR after switching to ALL induction therapy. Thus, the current study demonstrated that an ALL-directed induction chemotherapy is more effective in achieving CR than AML-type therapy, although patient numbers are small and follow-ups are not long. Rubnitz et al. [2] reported that ALL-type therapy was associated with higher CR rate than AML-type induction therapy (83% vs. 52%, respectively). Similar results were documented from studies by Matutes et al. [14] and by Al-Seraihy et al. [17]. In a Korean pediatric study, a significantly lower CR rate of 52% was reported even predominantly with ALL-type therapy. In this study, eight of ten patients who failed to achieve a CR with AML-type therapy subsequently entered a CR with ALL-type induction therapy [8]. Gerr et al. [15] reported significantly better survival in patients who received ALL-type induction therapy than those with AML-type induction therapy (81% vs. 41%, P=0.0009). In this study, same tendency was observed but they were not significantly different.
The issue of HSCT remains contentious. In this study, among 9 (75% of total cohort) patients who received an allogeneic HSCT, 5 (55.5%) survived. Among three patients who did not receive HSCT, only one survived. Our decision to proceed with transplant was primarily based on the availability of HLA-matched stem cell donor. The current study demonstrated a favorable outcome for patients transplanted in CR1. The post-allogeneic-HSCT outcome was much better than with post remission chemotherapy in a single institution (N=32, 3-year OS 77% vs. 16%) [28]. However, a Korean pediatric study reported no benefit from HSCT over chemotherapy alone in patients with BAL [8]. In general, similar outcomes have been reported between transplanted vs. non-transplanted cases, but the issues need to be clarified because of the lack of randomized study, due the small number and heterogeneity of patients, stem cell donor, and transplantation procedures. Thus, routine use of HSCT may not be required in patients with ALAL, especially in those who attain a molecular remission after induction therapy. However, still a significant portion of the patients may need a prompt HSCT. Some recommend that an HSCT should be reserved for the case of Philadelphia chromosome-positive MPAL, infants particularly those with 11q23 abnormalities and those who have a poor response to early therapy [2829]. However, whether older children with 11q23 abnormalities should also be transplanted remains unclear and require further evaluation using larger, possibly multi-institutional studies. Other risk-determining variables need to be included in this decision process [101229].
So far, the survival rate reported in both pediatric and adult studies ranged from 8.1% to 60% [21730]. In the current study, the 5-year OS of the total cohort was 51.1±15.8%. Most recently, the survival rate of MPAL based on the WHO classification was about 80% in children [16]. Killick et al. showed a better OS for the younger patients than older counterparts (75% vs. 17% at 2 yr; P=0.01) [7]. Gerr et al. [15] demonstrated that the 5-year EFS probability of ALAL patients (62±5%) was lower than those of ALL patients (80±1%, P<0.001), but better than those of AML patients (49±2%, P=0.027). A few studies demonstrated the survival of AUL or its comparison with MPAL, because of the extreme rarity of the cases. In the current study, the survival of MPAL (75.0±21.7%) was better than that of AUL (0.0%) (P=0.008) [30].
The prognostic implication of high leukocyte counts at diagnosis, and immunophenotypic subtype, expression of the specific antigen, as well as the selection of induction regimen and the use of HSCT in BAL or ALAL need to be further established in prospective, multicenter fashion. Pediatric ALAL may be distinct from its adult counterpart in terms of clinical and cytogenetic characteristics, and response to chemotherapy, even though they are extremely rare. With better understanding of novel mechanism and pathogenesis of these rare situations, the best therapeutic approaches including target therapy will be available in the future.


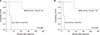
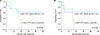









 XML Download
XML Download