

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Iron is a vital metal not only in hemoglobin synthesis but also in the structure of enzymes, cell growth and proliferation, the immune system, and electron transfer in body chemical interactions [1]. One of the most important causes of iron deficiency is gastrointestinal bleeding and menstruation in women [2]. One milliliter of packed RBCs contains one milligram of iron [3]. Diagnosis of iron deficiency anemia specifically in men is merely the beginning, as gastrointestinal study by endoscopy and colonoscopy must be performed for polyps, ulcers, and cancer [4]. Treatment of anemia resulting from bleeding requires sufficient iron resources for hematopoietic tissues.
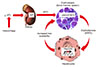
Erythroferrone
After bleeding, suppression of hepcidin gene expression causes an increase in gastrointestinal iron absorption and release of iron from cellular storage structures through the ferroportin channel [56]. In response to erythropoietin (EPO), hematopoietic tissue secretes erythroferrone (ERFE). Erythroferrone can rapidly suppress hepcidin gene expression for iron absorption and release of iron from the reticuloendothelial system (Fig. 1) [7]. Disorders of erythroferrone gene expression cause delayed increases in hemoglobin during bleeding and increased erythroferrone expression may lead to iron overload [5]. It is supposed that one of the reasons for iron overload in thalassemia syndromes is the increase in erythroferrone gene expression [8].
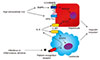
Hepcidin and inflammatory disorders
Iron metabolism
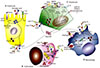
Systemic regulation of iron metabolism is presented in Fig. 3. In Fig. 3A, the absorption process through gastrointestinal cells in the duodenum is shown. Iron is imported through DMT1 (Divalent Metal Transporter) and exported through ferroportin [16]. Fig. 3B shows the iron transport to NRBCs (Nucleated red blood cells). Iron is transported by transferrin and upon binding to transferrin receptor (TFR), iron is released to the cytoplasm [1718]. The released iron is delivered to mitochondria by mitoferrin and the “Kiss and Run” process occurs, in which iron in endosomal vacuoles is released to mitochondria by direct contact with the mitochondrial membrane [19].
Fig. 3C shows the hepatocyte that controls iron absorption and release of iron from macrophages by regulating hepcidin secretion (Fig. 3D). Complexes of hemochromatosis proteins (HFE), hemojuvelin (HJV), matriptase-2, transferrin receptor 2 (TFR2), and BMP-6 (Bone morphogenetic protein) receptor on hepatocytes lead to hepcidin gene expression by activating SMAD and ERK-MAPK signaling pathways [202122232425]. Loss or gain of function mutations in these regulatory proteins leads to iron overload by suppressing hepcidin gene expression [26]. Reduction of hepcidin causes iron absorption from ferroportin [27]. GDF15 (growth differentiation factor 15), TWSG1 (twisted gastrulation 1), ERFE, and Matriptase 2 reduce hepcidin synthesis and increase iron absorption in the gastrointestinal system and release of iron from macrophages [28].
Role of microRNAs
Various microRNAs have a role in the expression of iron regulatory proteins. Increased or decreased microRNAs expression interferes in the translation process, causing an increase or decrease in translation and eventually altering the expression of certain proteins [29]. MicroRNAs are composed of about 21 nucleotides and lead to destruction and prevention of mRNA translation by hybridizing to them [30]. For instance, microRNA-130a expression is increased in iron deficiency and targets the mRNA of BMP receptor and causes reduction of hepcidin expression. In another example, reduction of microRNA-199a expression in response to hypoxia causes an increase of HIF-1α and HIF-2α expression that is subsequently accompanied by an increase in EPO synthesis [3132]. HIF factors (hypoxia inducible factors) are transcription factors for EPO gene expression [33].
Iron overload
The most common disorders of iron metabolism are iron overload (type I to IV), iron overload with cataracts, iron deficiency anemia, anemia of chronic disorders, and iron refractory iron deficiency anemia (IRIDA) [343536]. Mutations of hemochromatosis gene (HFE) or classic hemochromatosis appear in women after menopause and in 40–50-year-old men and are accompanied by iron overloads in heart, liver, and exocrine glands [3738]. Bronze skin, diabetes, and liver cirrhosis are complications of hemochromatosis that can lead to hepatocellular carcinoma [39]. Type II hemochromatosis, or juvenile type, is caused by mutations in the hepcidin gene (HAMP) or hemojuvelin (HJV) [4041].
Transferrin saturation ≥55% and ferritin levels ≥200 µg/L are the most important screen tests for hemochromatosis [42]. A certain type of hemochromatosis is accompanied by ferritin more than 1,000 µg/L and cataracts. In this situation, mutation in iron regulatory protein (IRP) prevents its binding to the iron response element of ferritin light chain mRNA so that suppression of ferritin synthesis does not occur [43].
Iron deficiency anemia
Iron deficiency anemia leads to reduction of serum iron, serum ferritin (SFt), and an increase in TIBC in progressive situations [44]. Since ferritin is an acute phase protein and increases in inflammatory and infectious processes, some physicians request CRP and ESR tests along with ferritin. The ferritin test is more reliable in negative CRP and ESR samples [45]. The first parameters of iron deficiency anemia are a reduction of CHR (reticulocyte hemoglobin) and an increase in RDW. Microcytic-hypochromia, an increase in RDW, and hypochromic pencil-shaped RBCs are observed in iron deficiency anemia when the anemia is in a progressive state and iron storage is depleted. In this situation, hemoglobin is below 10 (ferritin<10) and RBC count is <5 million per µL (Table 1) [46]. Iron deficiency anemia rapidly responds to iron therapy and an increase in reticulocyte count is typically observed after 5 days [47]. By beginning treatment, RBCs become dimorphic and populations of hypochromic and normochromic RBCs can be observed in blood smears and after some time, they are converted to normocytic-normochromic (Table 2) [48].
Within 3 weeks of treatment, hemoglobin typically increases to about 2 grams and when the amount of ferritin increases to 50, iron consumption can be stopped [49]. For measuring ferritin, it is not necessary to stop iron consumption and it can be measured any time of the day, whereas measurement of serum iron is suggested in the morning after fasting and daily alterations in serum iron (SFe) has been reported to be 30%. SFe is normal or increased in the morning and physiologically decreases in the evening. TIBC measurement is not highly affected by daily alterations. For measuring TIBC and SFe, iron consumption should be stopped for 2 or 3 days [50]. Any microcytic-hypochromic morphology that does not respond to iron therapy is classified into thalassemia syndromes, hemoglobinopathies, or IRIDA [51].
Iron refractory iron deficiency anemia
Iron refractory iron deficiency anemia can be inherited or acquired. A mutation in the TMPRSS6 gene that leads to disorders of matriptase-2 (MT2) synthesis is the most important inherited cause [5253]. In normal situations, MT2 inhibits the HJV protein's connection with the BMP-6 receptor and causes reduction of hepcidin gene expression [54], which is further accompanied by an increase in gastrointestinal iron absorption [55]. A mutation in MT2 causes an increase in hepcidin expression and destruction of ferroportin and thus iron absorption does not occur in this condition [56].
For children who do not respond to iron therapy despite the confirmation of iron deficiency, an anti-TTG test for diagnosing celiac disease, H. pylori infection, and autoimmune gastritis must be performed [57].
Pregnancy and iron status
In every pregnancy, according to the increased mass of red blood cells, needs of the fetus, and growth of placenta and delivery, the requirement of iron is increased [58]. Ferritin values less than 50 cause iron deficiency anemia in pregnant women, unless it is compensated with daily absorption of 3.5 mg of iron, and daily consumption of 20–30 mg of iron in a biocompatible form is sufficient for compensation [59].
Renal failure and iron deficiency
In renal failure patients who take EPO for treatment of anemia, the response to treatment can be predicted through transferrin saturation, free transferrin receptor in plasma (TFR), and ferritin levels [606162]. Transferrin saturation less than 20% with TFR >8 mg/L and ferritin <50 µg/L is a sign of iron deficiency and requires intravenous iron administration to increase ferritin levels to more than 100 µg/L for a sufficient response in EPO in patients not receiving dialysis, while sufficient ferritin levels for a response in EPO in dialysis patients are more than 200 µg/L [6364].
Megaloblastic anemia in accompaniment with iron deficiency anemia
Megaloblastic anemia exhibits characteristics of increased ovalomacrocytes (MCV>100), as well as increased RDW, MCH, and normal MCHC [68]. Megaloblastic anemia can be differentiated from cold agglutination by normal MCHC because in cold agglutination, MCV, MCH, and MCHC are increased [69].
Presence of hypersegmented neutrophils with ovalomacrocytes confirms the diagnosis of megaloblastic anemia [70]. If megaloblastic anemia is accompanied by iron deficiency anemia, it will cover macrocytic morphology and hypersegmented neutrophils will typically be observed in peripheral blood [71]. With vitamin B12 or folic acid treatment, microcytic-hypochromic morphology associated with iron deficiency anemia is observed [72].
Treatments
There are different drugs and methods in order to ameliorate the disorders which relate to iron metabolism. In iron deficiency anemia, oral iron is the first-line treatment, but in some conditions it is ineffective or harmful such as in inflammatory diseases and heavy bleeding. In these conditions, intravenous (IV) administration of iron is suggested and usually safe. Ferrous sulfate, gluconate, and fumarate are the most common oral iron formulations [7374]. In patients with non-dialysis-dependent chronic kidney disease and iron deficiency anemia, ferric citrate is effective and can correct anemia [75].
In hemochromatosis, venesection or phlebotomy, iron chelators and erythrocytapheresis are used for treatment but phlebotomy is the most acceptable method. In this method, the initial blood loss causes a reduction in hemoglobin stores of iron which helps erythropoiesis. It removes about 200–250 mg of iron in each session [7677].
In iron refractory iron deficiency anemia, patients do not respond to oral iron treatment appropriately but partial correction of anemia has been seen in some patients after a long period of oral iron administration [78].
CONCLUSION
Different factors play a role in molecular regulation of iron that maintain iron homeostasis by regulating the entrance and exit of iron. Defects or mutations in each of these factors can cause different clinical conditions related to iron metabolism. These disorders may have genetic backgrounds or can be generated by underlying diseases such as infection and inflammation. The most common disorders are iron deficiency anemia and anemia of chronic diseases, which can be diagnosed through hematological indices and genetic tests. Some patients respond to the usual treatments and some are resistant, especially those with IRIDA. Further studies are required to diagnose and treat these disorders.


 XML Download
XML Download