

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Genitopatellar syndrome (GPS) (MIM 606170) is a rare disorder that has been identified relatively recently. Patients present with multiple anomalies, including patellar hypoplasia, flexion contractures of the limbs, renal and genital anomalies, facial dysmorphism, microcephaly, agenesis of the corpus callosum, and intellectual disability.1 Since Cormier-Daire, et al.2 first described this entity as GPS and reported several cases of patients with these manifestations, <20 cases have been reported worldwide. The etiopathogenesis of GPS has been attributed to a mutation of the KAT6B gene, which was identified via whole exome sequencing (WES) in 2012.1 We report the case of a female infant who presented with typical clinical features of GPS in whom WES confirmed KAT6B mutation.
CASE REPORT
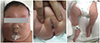
Our patient was a female infant born via cesarean section at 37 weeks' gestation to healthy unrelated parents. Antenatal ultrasonography showed unilateral multicystic dysplastic kidney (MCDK) and intrauterine growth restriction (estimated birth weight <10th percentile). At birth, her weight was 2210 g (3rd–5th percentile), height was 45.5 cm (5th–10th percentile), and head circumference measured 35.5 cm (75th–90th percentile). Her 1- and 5-min APGAR scores were 3 and 4, respectively. She was admitted to the neonatal intensive care unit after initial resuscitation with endotracheal intubation for the management of severe respiratory difficulty and poor muscle tone at birth. Physical examination showed light colored skin, a webbed neck with redundant skin, hypertelorism, small palpebral fissures, a broad nose, low-set and dysplastic auricles, a high palate, a small mouth, a hypoplastic gingival ridge, narrow chest contour, hypoplastic heels, and clitomegaly without any anal anomaly. We observed an abnormal posture with bilateral hip contractures, fixed flexion contracture of the right knee, and club feet (Fig. 1).
Renal ultrasonography performed after birth showed right MCDK and a small-sized left kidney. Renal failure with prolonged oliguria with consequent pulmonary edema and metabolic acidosis necessitated acute peritoneal dialysis from the 3rd to the 21st day of birth. She was subsequently administered erythropoietin and oral dichlozide to manage her chronic renal failure.
The patient received ventilator care including inhaled nitric oxide because of severe pulmonary hypertension until the 5th day of birth. Echocardiography showed large patent ductus arteriosus (PDA), an atrial septal defect, and a small muscular-type ventricular septal defect. Following improvement in her pulmonary hypertension, we performed PDA ligation to treat the hemodynamically significant left-to-right shunt through the PDA that had increased in size.
Brain ultrasonography showed partial agenesis of the corpus callosum. She developed seizures on the 10th day of birth, and electroencephalography showed occasional negative sharp waves across the bilateral temporal areas. Phenobarbital was administered for 2 months, and no further seizures occurred. Agenesis of the corpus callosum and delayed myelination was confirmed using brain magnetic resonance imaging on the 22nd day of birth.
Ventilator care was discontinued on the 30th day of birth, although she needed constant noninvasive ventilator care secondary to persistent hypotonia and poor respiratory drive. Laboratory investigations revealed primary hypothyroidism based on serum thyroid function tests (free thyroxine <0.4 ng/dL and thyroid stimulating hormone 213.62 µIU/mL) necessitating the administration of levothyroxine.
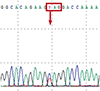
We performed WES to investigate the cause of the aforementioned multiple abnormalities and identified a heterozygous nonsense mutation in exon 18, p.Gln1515Ter, in the KAT6B gene (NM_012330.3: c4543C>T) (Fig. 2). This mutation has not been registered in the public database so far (Human Genetic Variation Browser, Exome Variant Server). Her karyotype was normal (46, XX). Direct Sanger sequencing verification of the KAT6B gene mutation performed in both parents revealed negative results, which confirmed this patient's de novo genetic mutation. She was finally diagnosed with GPS on the basis of clinical features and genetic testing.
The patient was discharged to her home after undergoing a tracheostomy at 6 months of age. She died at 8 months of age following respiratory arrest caused by tracheal tube dislocation at home.
This study was approved for exemption of subject consent by Severance Institutional Review Board (Assignment number 4-2018-1196).
DISCUSSION
Since being reported in 2000 on the basis of phenotypes,2 the molecular genetics of GPS were identified by Campeau, et al.1 and Simpson, et al.3 in 2012. The KAT6B gene located in the 10q22.2 region of DNA encodes lysine acetyltransferase 6B.1 KAT6B is an epigenetic regulator, and its mutation causes GPS and Say-Barber-Biesecker-Young-Simpson syndrome (SBBYSS).34 GPS and SBBYSS share several clinical features, including genital and patellar anomalies, hypotonia, congenital heart defects, hearing impairment, and intellectual disability.34 However, the two syndromes also show several features that differentiate one from the other. Patients with GPS primarily show musculoskeletal involvement of the lower extremities with patellar aplasia or hypoplasia, flexion contractures of the hips and knees, and club feet. In contrast, patients with SBBYSS typically show long thumbs and great toes.5 Genital anomalies that are characteristic of GPS are not a significant feature of SBBYSS. Patients with GPS usually do not show facial anomalies unlike patients with SBBYSS (mash-like facies, blepharophimosis, and ptosis).46 Mutations in the KAT6B gene causing GPS occur in the proximal segment of exon 18, whereas those causing SBBYSS appear to be clustered in exons 16–18.78
Our patient showed typical features of GPS without the long thumbs or lacrimal duct anomalies characteristic of SBBYSS (Table 1). Patellar hypoplasia could not be confirmed using imaging studies because patellar ossification usually occurs between 17 months and 4 years in girls.
The long-term prognosis of GPS is unknown and might depend upon the severity of associated organ dysfunction, including respiratory, cardiac, and renal failure. Our patient showed severe manifestations of GPS with bilateral renal anomalies and prolonged respiratory failure secondary to hypotonia.
GPS is a rare disorder and <20 cases have been reported worldwide. Thus, early diagnosis of GPS is difficult. In our patient we could identify multiple anomalies and promptly diagnose GPS using WES.
Kim, et al.9 reported the first case of GPS in South Korea in 2005 based on the patient's clinical phenotype. Our report is the first in South Korea to conclusively diagnose GPS based on genetic testing and would be a meaningful addition to the literature. In our view, this report will help clinicians to diagnose and predict the prognosis of this rare disorder.

 XML Download
XML Download