

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Non-small cell lung cancer (NSCLC) remains the leading cause of cancer-related death in men and women worldwide.1 Recent identification of druggable mutations and development of inhibitors targeting the alterations has improved the treatment outcomes of lung cancers.2345 However, in spite of the recent remarkable development of new treatment strategies, such as tyrosine kinase inhibitors and immunotherapies, patient survival has not markedly improved, suggesting the importance of discovering new genetic alterations and understanding their oncogenic roles on lung cancer.
In our previous study exploring the genome-wide profiles of chromosomal alterations in NSCLCs using microarray based comparative genomic hybridization (array-CGH) analysis, we observed that the 1q23.1 locus harboring the papillary renal cell carcinoma (PRCC) gene was recurrently amplified in NSCLC.6
PRCC, which encodes for a protein of 491 amino acids, is ubiquitously expressed in fetal tissues. PRCC has been reported as a fusion partner of a transcription factor TFE3 in some PRCC cases.789 Recent studies have shown that PRCC contains domains capable of transcriptional activation and that the fusion protein PRCC-TFE3 shows higher transcription activity than wild-type TFE3.7810 However, besides the fusion partner of TEF3, the oncogenic function of PRCC is almost unknown, because PRCC is not homologous to any known protein. PRCC is also reported to contain three proline-rich (P-X-X-P) motifs that can potently participate in binding to Src homology 3 (SH3) domain-containing proteins.7 Proteins with a SH3 domain are known to play an important role in signaling pathways regulating the cytoskeleton, Ras protein, and Src kinase.11 In addition, PRCC contains several potential sites for kinase phosphorylation, suggesting that PRCC is involved in a signaling cascade that may contribute to tumorigenesis.7 In support of this possibility, it was recently found that PRCC interacts with the cell cycle control protein Mad2B in renal cell carcinoma and translocates this protein to the nucleus where it exerts its mitotic checkpoint function.1213 These data suggest that overexpression of PRCC may contribute to the tumorigenesis of solid tumors including lung cancer through a mechanism different from fusion with TFE3. However, there has been no report on whether PRCC is overexpressed in NSCLCs or on the biological role of PRCC overexpression in lung tumorigenesis.
In this study, we aimed to explore the expression of PRCC in primary NSCLCs and the biological roles of PRCC overexpression on the tumorigenesis and progression of lung cancers by blocking the expression of PRCC in the human lung cancer cell lines harboring PRCC overexpression.
MATERIALS AND METHODS
Lung cancer cell lines
Human lung cancer cell lines (NCI-H23, NCI-H358, NCI-H460, and A549) were purchased from ATCC (American Type Culture Collection, Manassas, VA, USA) and maintained in DMEM and RPMI 1640 (Gibco BLR, Gaithersburg, MD, USA) supplemented with 10% FBS at 37℃ under 5% CO2. As a control, CCD-25LU (a human normal pulmonary epithelial cell line) was purchased from ATCC and maintained in Eagle's MEM supplemented with 10% FBS and 100 U/mL of penicillin/streptomycin.
Immunohistochemistry of NSCLC tissue microarray
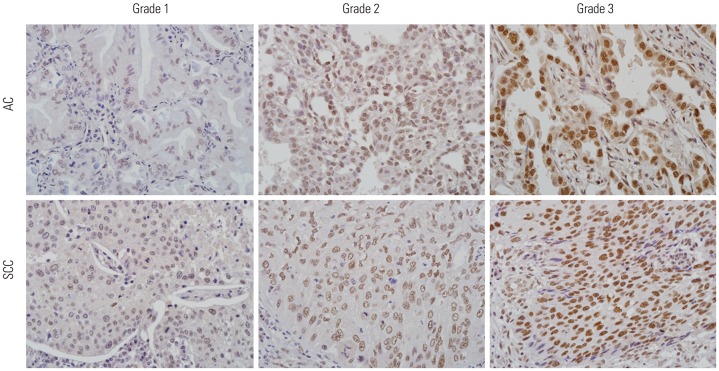
We used a lung cancer tissue microarray (TMA) developed at Seoul St. Mary's Hospital (Seoul, Korea) that contains 161 lung cancer tissues [81 adenocarcinomas (ACs) and 80 squamous cell carcinomas (SCCs)] under the approval of the Institutional Review Board of the Catholic University of Korea, College of Medicine (CUMC05U003). All cores from tumor tissue blocks were verified to contain tumor cells by histological examination. 4-µm sections of the TMA blocks were cut and used for immunohistochemistry (IHC) analysis. TMA sections were deparaffinized in xylene, hydrated with 100% ethanol and 95% ethanol, and rinsed in distilled water. Endogenous peroxidase was blocked with 0.1% H2O2. The section slides were then submitted to microwave antigen retrieval for pretreatment (10 mM citrate buffer, pH 6.0). The slides were incubated with serum blocking solution, primary antibody (anti-PRCC monoclonal antibody, clone D-3, 1:50, Santa Cruz Biotechnology, Santa Cruz, CA, USA), biotinylated secondary antibody, and streptavidin-horseradish peroxidase. Diaminobenzidine solution was used as a chromogen. The slides were counterstained in hematoxylin solution. The PRCC staining intensity was graded from 0 (no evidence of any nuclear immunoreactivity) to 3 (strongly positive immunoreactivity) by a board-certified pathologist. In this study, only the staining intensity of tumor cells was evaluated because the proportion of stained cells was constant throughout all cases. IHC grade 2 and grade 3 were deemed reflective of PRCC overexpression. Renal cell carcinoma and lung cancer tissues with known high expression of PRCC were used as a positive control for PRCC. The negative control used non-specific mouse IgG in place of the primary antibody.
Transfection of PRCC siRNAs
Three different PRCC-specific siRNAs (siPRCC-1, siPRCC-2, and siPRCC-3) were purchased from Invitrogen (Carlsbad, CA). Their sequences were as follows: siPRCC-1, UUG AUU UCU UCU CUC CCU CGG UUC CGGA ACC GAG GGA GAG AAG AAA UCA A; siPRCC-2, UGA CCA GGU GUU CUU CAG UUC CAG CGCU GGA ACU GAA GAA CAC CUG GUC A; siPRCC-3, AAG UCU UGG UCU UAG AAG CCA GUC UAGA CUG GCU UCU AAG ACC AAG ACU U. The siPRCC-1, -2, and -3 targeted exons 5, 7, and 3, respectively. To estimate the sequence-specific effectiveness of the PRCC-specific siRNAs, we also used a negative control siRNA (siNEG) (Invitrogen) that has no significant homology with any known sequences in the human genome. PRCC-specific siRNA was transfected into the cells at a final concentration of 100 nM using Lipofectamine RNAiMAX Transfection Reagent (Invitrogen) as described elsewhere.1415 Cells were harvested at different time points for the following tests.
Western blot analysis
Transfected cells were harvested and lysed in cell lysis buffer (50 mM NaF, 150 mM NaCl, 10 mM sodium pyrophosphate, 2 mM EDTA, 0.1% Triton X-100) with protease inhibitor. Cell lysate was electrophoresed on 10% SDS-polyacrylamide gel and the gels were blotted onto to a PVDF membrane (Millipore, Bedford, MA, USA). The membranes were blocked with 5% skim milk and then incubated overnight at 4℃ with anti-PRCC and anti α-tubulin antibodies (Sigma, St. Louis, MO, USA). Membranes were incubated with HRP-conjugated goat anti-rabbit IgG antibody (Sigma) for 1 hour at room temperature. Signals were visualized using an enhanced chemiluminescence system (Amersham-Pharmacia Biotech, Braunschweig, Germany). The expression of α-tubulin or β-actin was used as a control.
Antibodies
Antibodies for cyclin D1, AKT and phosphor-AKT were purchased from Cell Signaling Technology (Danvers, MA, USA). Antibodies for NFkB, CXCR-12, and vimentin were obtained from Santa Cruz Biotechnology. Ki67 antibody was purchased from Abcam (Cambridge, UK). Anti-rabbit, anti-mouse, and anti-goat secondary antibodies conjugated to HRP, FITC, and TRITC were obtained from Santa Cruz Biotechnology.
PRCC-specific real-time reverse transcription-PCR
Total RNA was isolated from cells using TRIzol reagent (Invitrogen). Reverse transcription was performed using oligo-(dT) primer and SuperScript® III reverse transcriptase (Invitrogen). Quantitative real-time reverse transcription-PCR (qRT-PCR) was performed with a ViiA™ 7 Real-Time PCR System using THUNDERBIRD™ SYBR® qPCR Mix (Toyobo, Osaka, Japan) and the following PRCC-specific primer set: 5′-ACACCTGGT CAGAGAACAAGCTCA-3′ (sense) and 5′-AGATCCTGGGAG CAATCAGTTCCA-3′ (antisense). GAPDH was used for normalization. Thermal cycling was performed as follows: one cycle at 95℃ for 10 min, followed by 45 cycles of 95℃ at 10 sec, 55℃ for 30 sec and 72℃ for 30 sec. Relative quantification was performed by the ΔΔCt method as described elsewhere.16
Cell proliferation assay
Cells were seeded onto 96-well plates (2×103 cells per well) and transfected with siPRCCs. Cell growth was determined by the cell proliferation reagent WST-1 (Roche, Indianapolis, IN, USA) according to the manufacturer's protocol. Optical density was read at 450 nm at various time points. Three independent experiments were done.
Invasion and migration assay
Invasiveness was assayed using the 24-well format transwell chambers (Becton Dickinson Labware, Franklin Lakes, NJ, USA) with 8 µm-pore size. siRNA transfected cells were added onto Matrigel (BD Biosciences, San Jose, CA, USA) coated filters in 200 µL of serum-free media (2×104 cells/filter). In the lower chambers, 500 µL of complete media containing 10% FBS was added. After 32-hour incubation at 37℃ in the 5% CO2 incubator, cells were stained with 0.5% crystal violet in 20% methanol and then examined under the microscope. Cells on the top surface of the membrane were removed by wiping with a cotton swab. The numbers of cells were counted in five microscopic fields at ×40 magnification. Migration assay was performed in the same manner but without the Matrigel coating on the filters. Six independent experiments were performed for each assay.
Colony formation assay
Forty-eight hours after siRNA transfection, cells were seeded into six-well plates. About one week later, cells were washed with PBS and stained with 0.5% crystal violet. The colonies were manually counted. Six independent experiments were performed for each assay.
Soft agar assay
Forty-eight hours after siRNA transfection, 1000 cells were plated onto six-well plates in complete culture medium containing 0.35% low-melting-point (LMP) agarose on top of 0.6% LMP agarose in the same medium. After incubating for 2 weeks at 37℃ in the 5% CO2, colonies >0.1 mm in diameter were counted under a microscopic field at ×40 magnification. Six independent experiments were performed for each assay.
Generation of enhanced green fluorescent protein stable expressing cell line
To generate the stably expressing enhanced green fluorescent protein (EGFP), pEGFP-N1 plasmid (2.5 µg) was transfected using Lipofectamine®. Forty-eight hours after transfection, cells were selected by 0.8 µg/mL of G418 (Sigma) treatment. After 10 to 14 days selection, the remaining colonies were isolated and maintained with 0.4 µg/mL of G418. EGFP expression was confirmed by fluorescence microscope and flow cytometry.
Mouse in vivo tumorigenesis model and intratumoral injection of siPRCC
Six-week old female BALB/c athymic (nude) mice were purchased from Charles River Laboratories (Milan, Italy) and maintained under specific pathogen-free conditions. EGFP stable-expressed NCI-H358 cells (H358-pEGFP-N1) were injected subcutaneously at both flanks (2×107 cells/mouse). When the average volume of each tumor, which was established from the injected EGFP stable cells, reached 50 mm3, siPRCC and siNEG were injected in 20 µL of PBS twice a week for 6 weeks. Tumor growth was monitored by measuring EGFP signals using the Maestro In Vivo Imaging System (Cambridge Research & Instrumentation, Inc., Woburn, MA, USA) with intra-peritoneal injection of ketamine/xylazine as the anesthetic drug combination. After 6 weeks, mice were sacrificed and tumor weights were measured. All animal experimental procedures were approved by The Catholic University of Korea Animal Ethics Committee (CUMS-2016-0251-03).
Statistical analysis
The statistical analysis was performed using the SPSS statistical software ver. 17 (SPSS Inc., Chicago, IL, USA). Statistical significance was determined by the two-tailed unpaired Student's t-test for continuous variables. p values less than 0.050 were considered statistically significant.
RESULTS
Expression of PRCC in human primary lung cancer tissues
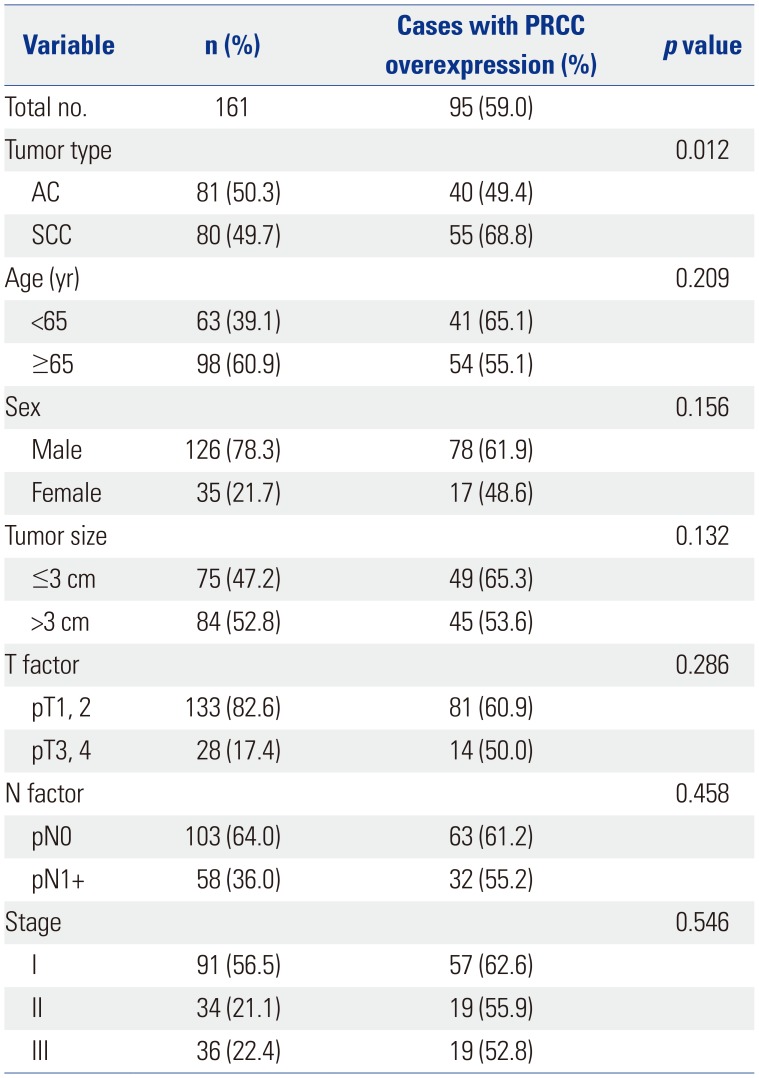
To examine the expression of PRCC in primary NSCLC, we screened PRCC protein expression level in 161 NSCLCs (81 ACs and 80 SCCs) by TMA-based IHC. Of 161 NSCLs, 95 (59%) showed PRCC overexpression, ≥IHC grade 2 (Table 1). Tumor cells showed nuclear expression of PRCC (Fig. 1). Normal lung tissue did not show PRCC overexpression (data not shown). PRCC overexpression was significantly more common in SCC than AC (p=0.012). Other factors, such as age, sex, tumor size, lymph node metastasis, and tumor stage, did not show significant correlation with PRCC expression status (Table 1).
Repression of PRCC expression by siPRCC in lung cancer cell lines
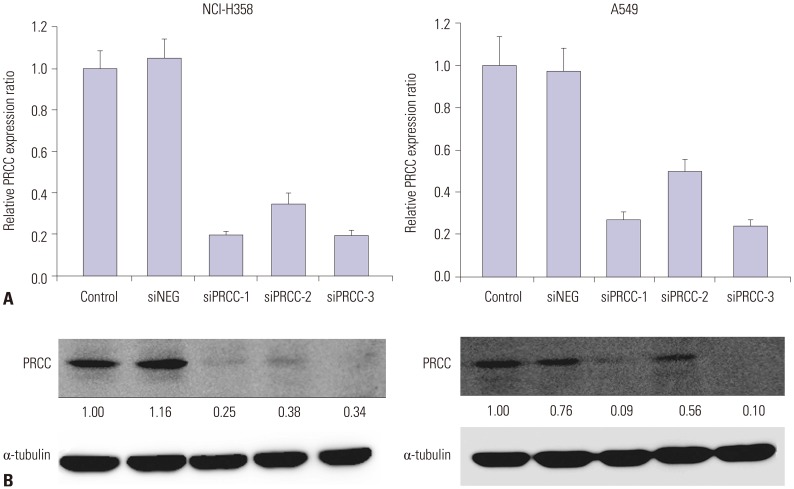
Before PRCC knockdown, we examined baseline PRCC expression levels in four lung cancer cell lines (NCI-H23, NCI-H358, NCI-H460 and A549) by Western blot analysis. In three of the four cell lines, protein expression levels of PRCC were profoundly increased, compared with normal lung cells (CCD25-LU) (Fig. 2). Among them, NCI-H358 and A549 showed the highest levels of baseline PRCC expression (4.51 and 3.87 folds, respectively); therefore, both cells were selected for the following experiments. To assess the biological effects of PRCC overexpression on lung carcinogenesis, we transfected three PRCC-specific siRNA constructs (siPRCC-1, -2, and -3) and siNEG into the NCI-H358 and A549 cell lines. Of the three siPRCCs, siPRCC-1 showed best knockdown performance in both cell lines (Fig. 3). Therefore, all downstream functional analyses were performed using the siPRCC-1 (hereafter called siPRCC).
The effects of PRCC silencing on lung cancer cell growth
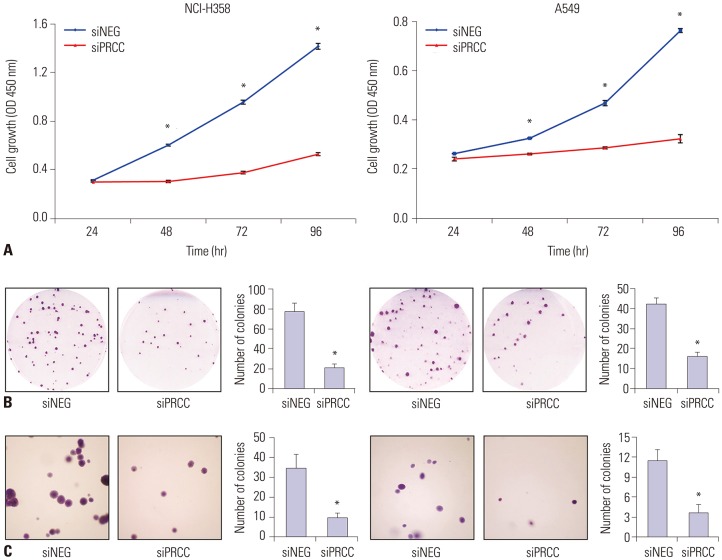
To explore the effects of PRCC on lung cancer cell growth, we performed proliferation and colony formation assays after PRCC knockdown. In the proliferation assay, both of the PRCC silenced cells showed significantly slower growth rates than that of the siNEG-transfected cells from 48 hours after siPRCC transfection (Fig. 4A). In the colony formation assay, the number of colonies from the siPRCC-treated cells was significantly lower than that from the siNEG-treated cells (NCI-H358: median 21.0, IQR 22.0–18.5 in siPRCC vs. median 77.0, IQR 83.0–72.5 in siNEG, p=1.902×10−8; A549: median 16.0, IQR 17.5–15.3 in siPRCC vs. median 42.5 in siNEG, IQR 43.8–40.5, p=4.467×10−9) (Fig. 4B). Next, we examined the PRCC-silencing effect on the anchorage independent growth of lung cancer cells by soft agar assay. The number of anchorage-independent colonies of the siPRCC-treated cells was significantly reduced, compared with that of the siNEG-treated cells (NCI-H358: median 10.0, IQR 11.8–7.5 in siPRCC vs. median 35.5, IQR 38.3–31.3 in siNEG, p=6.453×10−6; A549: median 3.5, IQR 4.8–3.0 in siPRCC vs. median 11.5, IQR 12.0–11.0 in siNEG, p=2.794×10−6) (Fig. 4C).
The effects of PRCC silencing on lung cancer cell migration and invasion
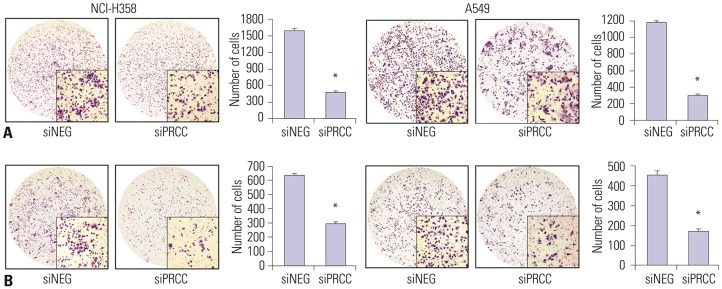
We next explored the effect of PRCC silencing on the capability of migration and invasiveness of the lung cancer cells. The number of migrating cells after PRCC knockdown was significantly lower than that in the siNEG-treated cells (NCI-H358: median 480.5, IQR 488.3–473.5 in siPRCC vs. median 1597.5, IQR 1618.8–1573.3 in siNEG, p=3.020×10−15; A549: median 298.5, IQR 308.0–292.8 in siPRCC vs. median 1171.5, IQR 1186.3–1162.8 in siNEG, p=4.152×10−16) (Fig. 5A). The number of invading cells was also significantly lower in PRCC knockdown than in the siNEG (NCI-H358: median 295.0, IQR 305.0–291.0 in siPRCC vs. median 641.0, IQR 645.5–626.0 in siNEG, p=4.532×10−13; A549: median 174.0, IQR 182.5–164.8 in siPRCC vs. median 457.0, IQR 464.8–445.5 in siNEG, p=1.633×10−11) (Fig. 5B).
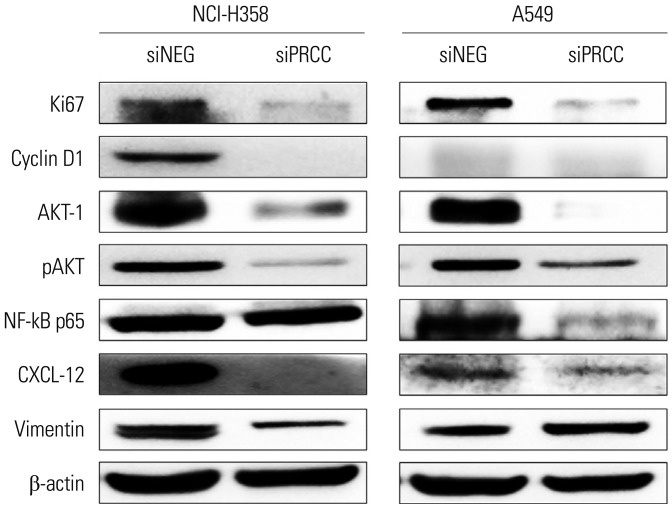
Mechanism of the anti-tumor effect after PRCC silencing
To explore the mechanism of tumor growth inhibition after PRCC silencing, we checked cell proliferation- and cell cycle-related markers, such as Ki67, cyclin D1, AKT-1, pAKT, and NF-kB p65 by Western blotting (Fig. 6). Expression levels of Ki67, AKT-1, and pAKT were profoundly decreased in both siPRCC-transfected cells, compared with siNEG-transfected cells. Expression levels of cyclin D1 and NF-kB p65 were also decreased after siPRCC treatment, but were observed in one of the two lung cancer cell lines: repressed cyclin D1 in NCI-H358 and repressed NF-kB p65 in A549. To explore the mechanism of repressed migration and invasion after PRCC silencing, we checked the expression of vimentin and CXCL-12 (Fig. 6). Expression of CXCL-12 was decreased in both siPRCC-transfected cells. Decreased expression of vimentin was observed in NCI-H358.
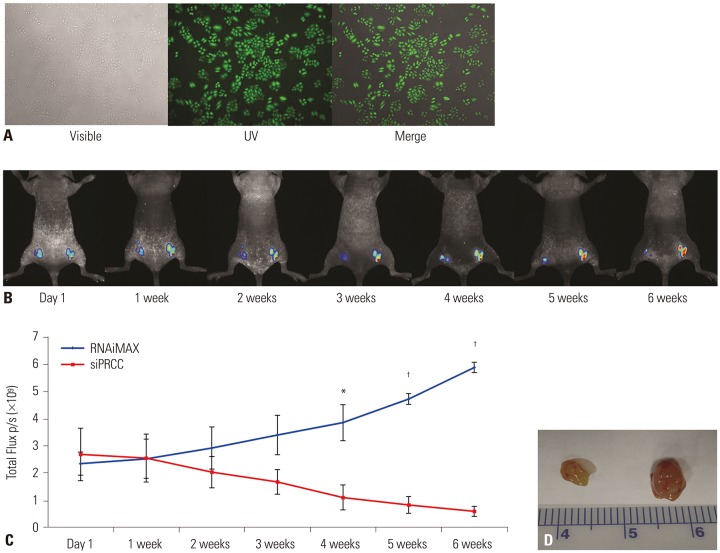
In vivo validation of anti-tumor effect after PRCC silencing
For visualization of in vivo growth of lung cancer cells, we constructed EGFP stable expressing NCI-H358 cells (H358-pEGFP-N1) (Fig. 7A). Then, the EGFP stable cells were injected into nude mice. To verify whether the growth suppression effect observed in in vitro PRCC silencing was consistently appeared in vivo, siPRCC was injected into the tumor mass for six weeks (twice a week), and we observed the tumor growth (Fig. 7B). After the initial intratumoral injection of siPRCC, the median fluorescence signal from the xenograft tumors was steadily reduced in time series observation up to 6 weeks (Fig. 7B and C). By contrast, the siNEG injected xenograft tumors showed steadily increased signals in time series observation. At 4 weeks after the injection, the fluorescence signals from the siPRCC-injected tumors became significantly lower than those from siNEG-injected tumors (p=3.837×10−5 at 6 weeks, n=6). When we collected the tumor mass after 6 weeks of initial injection, consistent with the growth kinetics, the weight of siPRCC-injected xenograft tumor became lower than that with siNEG-injected tumor (ratio of weights, 3.45; 95% CI, 172.50±129.83 in siNEG vs. 50.00±27.39 in siPRCC; p=0.070, n=6).
DISCUSSION
Copy number alteration is a common genetic event in solid tumors including lung cancer, and they are known to contribute to tumorigenesis by affecting the activities of cancer-related genes in the altered chromosomal regions.1718 Amplification of chromosome 1q has been reported to be recurrently occurred in NSCLCs.19 In our previous analysis of chromosomal alterations in NSCLC, the 1q21.1 locus was identified to be recurrently amplified.6 As this locus contains the PRCC gene, we speculated that over expression of PRCC may have an oncogenic role in lung tumorigenesis.
PRCC has been suggested as the fusion partner of TFE3 transcription factor, and the PRCC-TFE3 fusion protein showed higher TFE3 activity in renal cell carcinoma.78910 However, PRCC-TFE3 fusion has not been reported in other solid tumors, suggesting that PRCC itself may have a different oncogenic mechanism in other solid tumors. Indeed, PRCC is known to contain proline rich motifs that can bind to SH3 domains.7 This interaction can play a role in regulating the cytoskeleton, Ras protein, and Src kinase pathway.11 This situation led us to investigate the role of PRCC in NSCLC. The aim of this study was three-fold. First, we aimed to identify the profiles of PRCC expression in Korean NSCLC patients. Second, we aimed to elucidate the role of PRCC overexpression in lung tumorigenesis by knocking down overexpressed PRCC in two lung cancer cell lines that showed PRCC overexpression. Third, we attempted to confirm the in vitro effect of PRCC silencing observed in tissue culture condition by in vivo mouse model.
In IHC analysis of 161 Korean NSCLC cases, we found that 59% of the lung cancers involved PRCC overexpression. To our knowledge, this is the first evidence showing that overexpression of PRCC is a common occurrence in lung cancer. PRCC overexpression was significantly more common in SCCs than ACs; however, we could not observe significant correlations with other clinical features, such as tumor size, lymph node metastasis, or tumor stage, in this study. Considering that we analyzed a limited number of NSCLC cases in this study, further study will be required to confirm the clinical meaning of PRCC. The SCC dominance of PRCC overexpression observed in this study also needs further confirmation in a larger cohort. Although we could not make conclusive interpretation of PRCC overexpression on the clinical features of NSCLCs, PRCC overexpression in approximately 60% of the primary NSCLCs strongly suggests that PRCC overexpression can be useful as a tumor marker of NSCLC and may be involved in lung tumorigenesis.
In addition to the primary NSCLCs, three of the four lung cancer cell lines showed highly overexpressed PRCC, further supporting its oncogenic potential. After silencing the PRCC in two PRCC overexpressed lung cancer cell lines, cell growth rate, colony formation, anchorage independent growth, and invasiveness were repressed in both cells. Although we did not perform the induction of PRCC overexpression in PRCC non-overexpressed, normal lung epithelial cells to induce lung tumorigenesis, our data supports the role of PRCC overexpression in NSCLC tumorigenesis.
To determine the molecular mechanism of tumor growth inhibition after PRCC silencing, we assessed cell proliferation-related markers and compared the expression of molecules between siPRCC- and siNEG-treated cells. As expected, key molecules known to be involved in cell proliferation and cell cycle, such as Ki67, cyclin D1, AKT-1, pAKT, and NF-kB p65, were decreased in siPRCC-transfected cells, compared with siNEG-transfected cells, suggesting that overexpression of PRCC is involved in uncontrolled cell proliferation, which may contribute to lung tumorigenesis. In addition to the repressed cell proliferation, suppression of invasiveness was also observed after PRCC silencing in this study. To understand the molecular mechanism of repressed migration and invasion after PRCC silencing, we evaluated the expression of vimentin and CXCL-12. Vimentin is one of the key EMT markers that can induce tumor metastasis.20 Chemokines are known to play important roles in metastasis of cancer cells, and CXCL-12 is known to be involved in regulation of metastasis. Moreover, overexpression of CXCL-12 has been reported in various metastatic solid tumors, including lung cancer.21 In this study, expressions of related molecules were decreased after PRCC silencing, suggesting that overexpression of PRCC is involved in overexpression of vimentin and CXCL-12, which may contribute to enhancing the metastatic potential of lung cancer.
To trace the in vivo effects of PRCC silencing, we developed EGFP stable lung cancer cells and xenografted the cells into nude mice. When we injected siPRCC into the xenograft tumors, tumor growth was significantly reduced, compared with siNEG-injected tumors. Through this experiment, we confirmed the in vivo effect of PRCC silencing. All of the PRCC IHC data with primary NSCLC tissue array and in vitro/in vivo data suggest that PRCC overexpression can contribute to lung tumorigenesis. Additionally, repression of tumor growth after siPRCC injection into the mouse xenograft tumor also suggests the possibility that PRCC can be a therapeutic target.
There are several limitations to this study. First, although we observed recurrent PRCC overexpression in the primary NSCLCs and growth/migration inhibition after silencing overexpression in vitro, we could not observe significant clinical associations other than squamous cell carcinoma dominance in our primary NSCLCs. This may be due to the limitation of NSCLC cases in the NSCLC tissue array used in this study. Further larger scale analyses will be required to attain conclusive insights into the clinical implications of PRCC overexpression. Second, we did not evaluate whether artificial induction of PRCC overexpression can induce lung tumorigenesis in PRCC non-overexpressed normal lung epithelial cells.
In conclusion, we observed that PRCC overexpression is a common event in NSCLCs. When we silenced PRCC expression in PRCC overexpressed cells, tumor growth was significantly reduced in vitro and in vivo. These data suggest that PRCC overexpression is involved in tumorigenesis and progression of lung cancer. This study provides a better understanding of lung tumorigenesis and suggests PRCC as a potential biomarker and therapeutic target of lung cancer.

 XML Download
XML Download