

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Benign recurrent intrahepatic cholestasis (BRIC) is defined and characterized by intermittent episodes of cholestasis without progression to liver failure. These episodes include symptoms of pruritus and jaundice without extrahepatic bile duct obstruction [1]. The exact prevalence of BRIC remains unknown, but its estimated incidence is approximately 1 in 50,000 to 100,000 people worldwide [2]. BRIC, a rare autosomal recessive inherited disorder in children, is divided into two types, BRIC1 and BRIC2, according to its genetic cause. An ATP8B1 gene mutation causes BRIC1, while an ABCB11 gene mutation causes BRIC2, but their phenotypes are nearly identical. A 2004 publication was the first report of a mutation in ABCB11, the gene encoding the hepatocellular bile salt export pump (BSEP), causing BRIC2. Mutations in ABCB11 are also present in progressive familial intrahepatic cholestasis type 2 (PFIC2), but the phenotypes of PFIC2 and BRIC2 differ, despite both being caused by mutations in the same gene [3]. PFIC2 is characterized by progressive liver damage, is more severe than BRIC, and usually requires liver transplantation [4]. Here we report the first case of Korean BRIC2 siblings with novel BSEP mutations.
CASE REPORT
A 6-year-old girl had recurrent episodes of jaundice and pruritus. At two months of age, jaundice with hepatosplenomegaly occurred for the first time. There was no significant familial or medication history. Liver function tests showed cholestatic hepatitis, including elevated levels of bilirubin and liver enzymes: serum aspartate aminotransferase (AST), 2,122 IU/L; alanine aminotransferase (ALT), 1,291 IU/L; total bilirubin (TB), 18.3 mg/dL; and direct bilirubin (DB), 11.0 mg/dL. The testing also revealed a low γ-glutamyltransferase (GGT) level (59 IU/L). The neonatal screening test results for inherited metabolic diseases were normal and other biochemical parameters were within normal ranges. Viral and parasite studies were negative for hepatitis A, B, and C viruses; cytomegalovirus; Epstein-Barr virus; herpes simplex virus; rubella virus; and toxoplasmosis.
On examination, her abdomen was soft and slightly rigid. Bowel sounds were normoactive, and the liver and spleen were both palpated two fingerbreadths below the costal margin. Ultrasonography of the abdomen revealed hepatosplenomegaly, a diffuse gall bladder wall, periportal edema, and no evidence of biliary atresia. A liver biopsy showed a diffuse giant cell transformation, ballooning degeneration, bile duct paucity, intracytoplasmic cholestasis, moderate portal inflammation, and periportal fibrosis (Fig. 1A).
 | Fig. 1(A) Photomicrograph of the liver biopsy shows diffuse giant cell transformation of hepatocyte, ballooning degeneration, bile duct paucity, moderate portal inflammation and intracytoplasmic cholestasis (initial liver biopsy, H&E stained, ×400), and (B) intracanalicular cholestasis, bile duct paucity, and minimal to mild focal pericellular fibrosis (recurrent state follow up liver biopsy, H&E stained, ×400).H&E: hematoxylin and eosin.
|
She was given medium-chain triglyceride formula, vitamin D, ursodeoxycholic acid, tocopherol, and multivitamins. Her liver function test results were improved a week later: AST, 536 IU/L; ALT, 369 IU/L; TB, 7.8 mg/dL; DB, 6.1 mg/dL; and GGT, 44 IU/L.
ABCB11 and ATP8B1 gene studies were performed, and the latter was normal. However, the ABCB11 gene study revealed novel compound heterozygous mutations, including p.R1221K and c.2075+3A>G in IVS17. A familial gene study showed p.R1221K heterozygote status in the mother and c.2075+3A>G in IVS17 heterozygote status in the father. There was no family history except her brother (Fig. 2A).
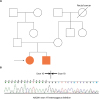 | Fig. 2(A) Pedigrees of family who were subjected to familial genetic analysis and (B) reverse transcription polymerase chain reactions of ABCB11 gene in both patients showed exon 17 heterozygous deletion.
|
The guanine was replaced by adenosine (c.3662G>A), leading to an amino acid substitution from arginine to lysine (p.R1221K) at the protein level. This mutation is found in 0% (0/180 alleles) of normal controls. The effect of the amino acid substitution on protein function was predicted by PolyPhen-2 analysis (http://genetics.bwh.harvard.edu/pph2) and the result was predicted to be likely damaging [5]. The pathogenic effects of the missense mutation were also evaluated by Mutation Taster (http://www.mutationtaster.org), which revealed its potential to cause disease [6]. The splicing site mutation of c.2075+3A>G is located at the site of intron 17 and causes exon 17 skipping. Exon 17 deletion in our patient was confirmed by reverse transcription polymerase chain reaction (Fig. 2B).
Her liver function test results were normal at 12 months of age and she was managed without any medication. However, at six years of age, abdominal pain, steatorrhea, jaundice, and pruritus recurred. Jaundice and pruritus were aggravated and the abdominal pain was relieved after yellowish diarrhea. Her total and direct bilirubin levels were elevated again (TB, 17.0 mg/dL; DB, 11.92 mg/dL), while other liver enzyme levels were normal or slightly higher than normal (AST, 58 IU/L; ALT, 36 IU/L; GGT, 14 IU/L). After treatment with rifampin and phenylbutyrate, the bilirubin values improved (TB, 12.5 mg/dL; DB, 9.18 mg/dL). The pruritus and steatorrhea recurred and the liver enzyme levels increased again when the medication was tapered (TB, 17.4 mg/dL; DB, 12.96 mg/dL; AST, 91 IU/L; ALT, 51 IU/L; GGT, 32 IU/L). A repeat liver biopsy showed bile duct paucity, slender periportal fibrosis, mild pericellular fibrosis, and intracanalicular cholestasis, but the fibrosis had not progressed (Fig. 1B). The liver function test results and pruritus improved with phenylbutyrate and rifampicin treatment. Following the last episode and medication cessation, she had no symptom recurrence. BRIC2 was diagnosed by the clinical findings of recurrent cholestasis and the genetic finding of an ABCB11 mutation.
Our patient's younger brother was three years old and had jaundice at two months of age. He had loose stools and no vomiting and his jaundice lasted for one month. An evaluation for neonatal cholestasis was performed and his liver function tests also showed cholestatic hepatitis and a low GGT level (AST, 356 IU/L; ALT, 538 IU/L; TB, 5.1 mg/dL; DB, 3.82 mg/dL; GGT, 43 IU/L). Metabolic disease screening test results were within the normal range. Abdominal sonography showed no hepatomegaly, revealed normal hepatic parenchymal echogenicity, and excluded biliary atresia. Genetic testing revealed the same mutations as our patient, his sibling. His liver function test results were normal at six months of age and have remained normal since.
DISCUSSION
BRIC is characterized by recurrent episodes of cholestasis without progressive liver disease. The clinical features of BRIC were first described in 1959 [7], and Luketic and Shiffman [8] have since proposed the following diagnostic criteria: 1) at least two episodes of jaundice separated by a symptom-free interval lasting several months to years, 2) laboratory data consistent with intrahepatic cholestasis, 3) a normal or minimally-elevated GGT level, 4) severe pruritus secondary to cholestasis, 5) centrilobular cholestasis evident on a liver biopsy, 6) normal intra- and extrahepatic bile ducts on cholangiography, and 7) an absence of factors known to be associated with cholestasis. Our patients fulfilled these criteria.
BRIC2 and PFIC2 are considered to be part of a spectrum of intrahepatic cholestasis disorders and the difference between the two diseases is based on phenotypic presentation. They have the same genetic mutation in ABCB11, the gene encoding the BSEP protein, the transporter for the secretion of bile salts from the hepatocytes into the canaliculus [3]. The BSEP is the most important transporter of bile salts into the canalicular space, so a deficiency in this gene can cause severe cholestasis. ABCB11 gene mutations reduce BSEP function and eventually reduce bile salt secretion, which causes the features of BRIC2. More than 100 ABCB11 mutations have been identified and more than half are missense mutations, including E297G or D482G, the most common BRIC-associated mutations in European families [9,10]. Genetic investigation of the corresponding genes can confirm this diagnosis and immunohistochemical analysis of BSEP in the liver tissue of patients with an ABCB11 deficiency shows reduced or absent BSEP staining [10,11]. Compared to BRIC, about half of the PFIC2 mutations involve an early stop codon or frameshift in the encoded protein [12].
By sequencing all coding exons of ABCB11 and ATP8B1, we detected c.2075+3A>G and p.R1221K in the ABCB11 gene of both siblings. The p.R1221K mutation has not been reported before but the effect of this mutation can be predicted by PolyPhen-2 and Mutation Taster. This mutation was expected to affect protein structure and host phenotype. The p.R1221K mutation in the ABCB11 gene is likely to be pathogenic by the criteria for classifying variants in the American College of Medical Genetics and Genomics guidelines [13]. Our patient had a splicing-site mutation of c.2075+3A>G, which is located at intron 17. This splicing mutation led to exon 17 skipping. A BRIC2 patient was previously reported to have a splicing-site mutation, a nucleotide alteration of IVS 19+1 G>A. Furthermore, 15 splicing site mutations of ABCB11 were identified in PFIC2, as well as in BRIC2 patients [3,10]. Exon 17 skipping was also reported in a PFIC patient with an IVS 16-8T>G mutation [14]. Therefore, BRIC2 was confirmed by discovering heterozygotic novel mutations in the ABCB11 gene.
Treatment of BRIC is symptomatic. No specific treatment to prevent attacks or limit their duration is available [8]. Medication therapy for BRIC involves relieving symptoms, such as pruritus, for which rifampicin and phenylbutyrate are effective [8,15]. Pruritus is caused by the accumulation of bile acid in the hepatocyte membranes. Rifampin competes with the hepatic uptake of bile acid, thus lowering hepatocyte bile concentrations and reducing pruritus [16]. Phenylbutyrate, histone deacetylase inhibitor 4-phenyl butyric acid, was reported to increase functional BSEP expression and bile acid transport via the canalicular membrane in vivo in E297G and D482G BSEP, common missense mutations in European BRIC2 patients [15,17,18]. Additionally, its effect on the biochemical markers of cholestasis and liver histology likely depend on the mutation type [19]. Phenylbutyrate was an effective drug for treating various mutations, such as PFIC2 and BRIC2 in in vitro studies. In particular, it decreased the biological parameters of cholestasis, increased biliary bile acid secretion, and partially corrected BSEP canalicular expression detected by immunostaining in PFIC2 patients with ABCB11 mutants [20]. Our patient responded well to phenylbutyrate and rifampicin. However, a further in vitro study is needed to evaluate the effect of phenylbutyrate on the transport function of novel BSEP mutations and to develop mutation-targeted therapy.
In conclusion, this is the first report of BRIC2 confirmed by an ABCB11 gene study in Korean siblings. The novel identification of the mutations of c.2075+3A>G in IVS17 and p.R1221K could help further our understanding of the mechanism of BRIC and determine mutation-specific therapies in the future.
 XML Download
XML Download