

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Current therapeutic strategies for acute leukemia include induction chemotherapy and additional consolidation or maintenance chemotherapy using cytotoxic agents; however, patients face a high risk of mortality or morbidity during the chemotherapy cycle. Monoclonal antibodies against leukemia-associated antigens induce antibody-dependent cellular cytotoxicity by recruiting tumor cells and exert cytotoxicity via binding to activated immune effector cells. These antibodies could provide an alternative therapeutic strategy to chemotherapy [123456].
The JL1 antigen is an epitope of CD43, a cell surface glycoprotein of the mucin family. It is also known as a differentiation antigen expressed on double positive (CD4+ CD8+) human cortical thymocytes [6789]. In normal bone marrow (BM), the JL1 antigen is expressed neither on hematopoietic pluripotent stem cells of myelomonocytic and erythroid lineages nor on mature blood cells (lymphocytes and segmented neutrophils) [789]. In contrast, the JL1 antigen is heterogeneously expressed in myelomonocytic lineages (no or weak expression on promyelocytes, myelocytes, and metamyelocytes, but strong expression on band forms); in lymphoid lineages, JL1 antigen is expressed on common lymphoid progenitors and widely distributed during lymphoid maturation but is down-regulated during the final maturation process [789]. The JL1 antigen has also been reported to be expressed on leukemic T, B, and myeloid lineage cells in >80% of acute leukemia patients and thus could serve as a potential candidate for immunotherapy [789]. The overall incidence of JL1 positivity was reported to be 80.9%, 87.0%, and 90.1% in non-T ALL, T-ALL, and AML, respectively [7]. Preclinical studies have demonstrated the cytotoxic effects of an anti-JL1-based immunotoxin against JL1-positive leukemic cells, which does not affect most normal tissues other than thymocytes and certain mononuclear cells of the BM [10]. A study reported that the JL1 monoclonal antibody recognizes a specific epitope of human CD43 and that saporin conjugated to the JL1 antibody can effectively attack leukemic cells in in vitro cytotoxic assays and result in significantly prolonged survival in a leukemic mouse model [11].
Effective immunotherapy for acute leukemias depends on the exclusive expression of an antigen on leukemic blasts but not on normal hematopoietic cells [12]. Anti-CD20 monoclonal antibody rituximab for B-lineage lymphoma demonstrated the rationale for using immunotherapy in hematologic malignancies [13141516]. Previous studies focused on the treatment of refractory or high-risk acute leukemias. To date, no comprehensive clinical, immunophenotypic, and genetic investigation of JL1-expressing de novo pediatric acute leukemias has been performed. We investigated the incidence of JL1 expression and compared the clinical, immunophenotypic, and genetic characteristics of de novo pediatric acute leukemia patients with respect to JL1 expression status to determine the therapeutic potential of an anti-JL1 monoclonal antibody.
METHODS
Patient cohort and acquisition of clinical and prognostic data
In total, 82 patients with pediatric acute leukemia (52 ALL [48 B-ALL and four T-ALL] and 30 AML) diagnosed between December 2014 and January 2016 at Asan Medical Center, Seoul, Korea, were initially enrolled. Four AML patients who were identified as having secondary AML, such as therapy-related AML or AML with myelodysplasia-related changes, were subsequently excluded. We finally included 78 patients diagnosed as having de novo pediatric acute leukemia (52 ALL and 26 AML) with a median age of 96 months (range: 2–216 months) and a median follow-up period of 424 days (range: 79–753 days). This prospective study was conducted in accordance with the Declaration of Helsinki (2013 revision), and written informed consent was obtained from all patients. It was approved by the Institutional Review Board (IRB) of Asan Medical Center, Seoul, Korea (approval number: AMC IRB 2014-0066).
Clinical characteristics, including sex, age, hemogram results, and blast proportions in peripheral blood (PB) and BM at diagnosis, were collected. The prognostic indicator period from diagnosis to first hematologic complete remission (CR), rate of hematologic CR achievement, and relapse rate in patients with first hematologic CR were analyzed. Hematologic CR was defined as meeting all of the following response criteria during evaluation at least four weeks post diagnosis: <5% blasts in the BM and no blasts in the PB, normal maturation of all cellular components in the BM, no evidence of extramedullary diseases (such as involvement of the central nervous system or soft tissue), absolute neutrophil count ≥1×109/L, platelet count ≥100×109/L, and a clinically proven transfusion independent state. Relapse was defined as the presence of ≥5% leukemic blasts in the BM aspirates in patients with a previously hematologic CR state.
Treatment strategies
Patients diagnosed as having ALL received induction chemotherapy comprising vincristine, corticosteroids, and L-asparaginase with added anthracycline. In addition, intrathecal chemotherapy was administered with methotrexate for standard-risk patients and with corticosteroid and cytarabine for high-risk patients, such as those with T-ALL or a complex karyotype. Following induction chemotherapy and achievement of hematologic CR, consolidation chemotherapy typically lasting one to two months was conducted with methotrexate and 6-mercaptopurine or 6-thioguanine for standard-risk patients; L-asparaginase, doxorubicin, etoposide, cyclophosphamide, and cytarabine for high-risk patients; and a tyrosine kinase inhibitor for patients with t(9;22)(q34;q11.2). Following consolidation chemotherapy, maintenance chemotherapy typically lasting 16 weeks was performed; the most commonly applied treatment regimen was administration of 6-mercaptopurine daily and methotrexate weekly, administered as pills, often together with vincristine (administered intravenously) and a corticosteroid (administered orally). The latter two drugs were administered once after every four to eight weeks.
Patients diagnosed as having AML received induction chemotherapy (7+3 regimen) with a continuous infusion of cytarabine at a dosage of 100–200 mg/m2 per day on days 1 to 7 and daunorubicin at 60 mg/m2 per day on days 1 to 3. Following hematologic CR, patients received high-dose cytarabine-based consolidation chemotherapy using cytarabine twice daily at a 3 g/m2 dose on days 1, 3, and 5, or received hematopoietic stem cell transplantation according to the eligibility of hematopoietic stem cell donors. In cases of acute promyelocytic leukemia, patients received induction chemotherapy consisting of all-trans retinoic acid (ATRA) plus daunorubicin and/or cytarabine, as well as consolidation chemotherapy with similar regimens.
Immunophenotyping
Flow cytometry for immunophenotyping leukemic blasts was performed with a FACSCanto II flow cytometer (BD Biosciences, San Jose, CA, USA). For ALL, CD45, CD34, terminal deoxynucleotidyl transferase (TdT), CD13, CD33, CD10, CD19, CD20, cytoplasmic CD22 (cCD22), CD2, CD3, cytoplasmic CD3 (cCD3), CD5, CD7, CD56, cytoplasmic IgM (cIgM), surface IgM (sIgM), and myeloperoxidase (MPO) were included in the analysis. For AML, CD45, CD34, TdT, CD13, CD33, CD10, CD19, cCD22, CD2, CD3, cCD3, CD7, CD56, MPO, HLA-D related (HLA-DR), CD117, CD65, CD15, CD14, and CD41 were included in the analysis.
Expression of JL1 on the leukemic blasts was also examined using FACSCanto II. At least 20,000 nucleated cells were acquired per tube, and leukemic blasts were isolated based on weak to intermediate expression of CD45-allophycocyanin (APC)/low side scattering (SSC) gating. The proportion of JL1 expressing leukemic blasts among the gated leukemic blasts was calculated based on the forward scattering (FSC) vs JL1-phycoerythrin (PE) dot plot. The results of the JL1 expression analysis are presented as the proportion of JL1-positive cells among the gated leukemic blasts (Fig. 1). The cutoff threshold of the fluorescence intensity for positive JL1 expression was initially determined using isotypic controls, and its validity was finally confirmed using an internal control (lymphocytes without JL1 expression) for each analysis. Positive JL1 expression was defined as ≥20% expression among the gated leukemic blasts. All antibodies except the ones for CD45 (BD Biosciences) and JL1 (Dinona Inc., Seoul, Korea) were provided by Beckman-Coulter (Miami, FL, USA).
Genetic analysis
Conventional karyotype analysis was performed for all patients at diagnosis. HemaVision (DNA Diagnostic, Risskov, Denmark) PCR and direct sequencing for the detection of fms-related tyrosine kinase 3 gene internal tandem duplications (FLT3-ITD), FLT3-tyrosine kinase domain (TKD), nucleophosmin (NPM1), c-KIT, and CCAAT/enhancer-binding protein-α (CEBPA) mutations were also performed at diagnosis. In addition, the proportion of recurrent genetic abnormalities (RGA), defined in the 2016 WHO classification, were calculated [17].
Statistical analysis
The Kolmogorov-Smirnov test was performed to assess the normality of the distribution of continuous variables. Normally distributed continuous variables were summarized as mean and SD and compared using the Student t-test. Non-normally distributed continuous variables were summarized as median and range and compared using the Mann-Whitney U test. Dichotomous variables were summarized as number and percentage and compared using Pearson's chi-square test and Fisher's exact test (for N<5). All tests were two-tailed. P≤0.05 was considered statistically significant. All tests were performed using SPSS for Windows, version 21.0 (IBM Corp., Armonk, NY, USA).
RESULTS
Comparison by disease subgroups
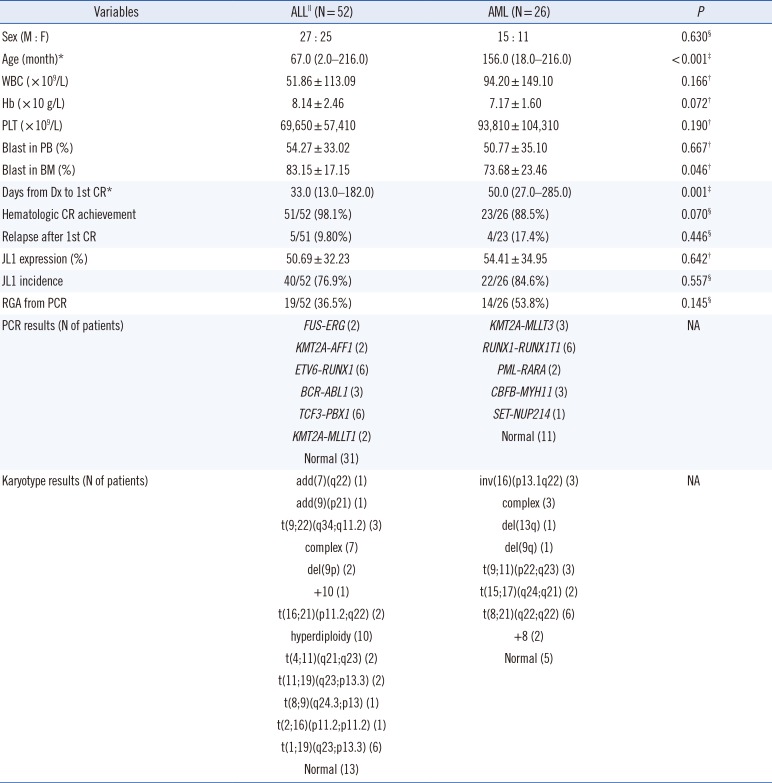
ALL patients were younger than AML patients at diagnosis (median 67 vs 156 months, P<0.001), and their periods from diagnosis to achievement of first hematologic CR were shorter (median 33 vs 50 days, P=0.001). However, no other clinical variables differed significantly between ALL and AML patients, nor did JL1 expression in leukemic blasts and JL1 incidence (mean 50.69% vs 54.41%, P=0.642 and 76.9% vs 84.6%, P=0.557, respectively; Table 1).
ALL patients
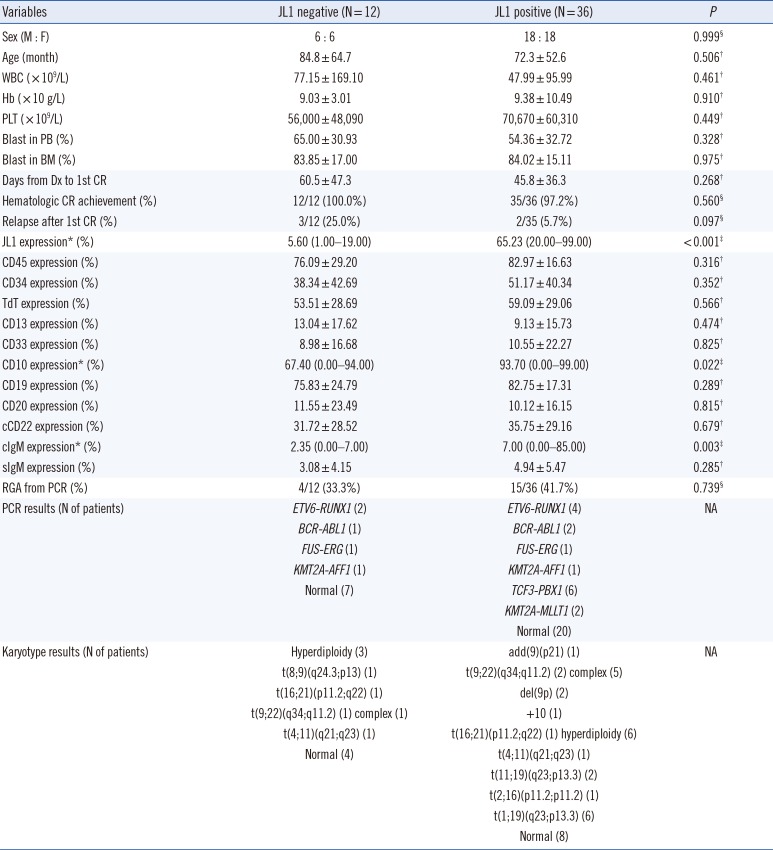
The clinical characteristics, including prognosis, of the 48 B-ALL patients did not significantly differ with respect to the JL1 expression status. The patients with positive JL1 expression showed higher JL1 expression in leukemic blasts than those without JL1 expression (P<0.001). Notably, the patients with positive JL1 expression showed higher CD10 and cIgM expressions in leukemic blasts than those without JL1 expression (P=0.022 and 0.003, respectively).
The patients with positive JL1 expression also showed a high incidence of TCF3-PBX1 (N=6) and KMT2A-MLLT1 (N=2) rearrangement (detected by PCR), indicating the presence of t(1;19)(q23;p13.3) and t(11;19)(q23;p13.3) chromosomal aberrations, respectively. The presence of these two chromosomal aberrations in eight patients was further confirmed by karyotype analysis. These two genetic abnormalities were not observed in patients without JL1 expression (Table 2).
Four T-ALL patients exhibited JL1 expression (median, 45.0%; range, 31.6–88.2%), and their karyotype was normal, complex, hyperdiploidy, and add(7)(q22), respectively (data not shown).
AML patients
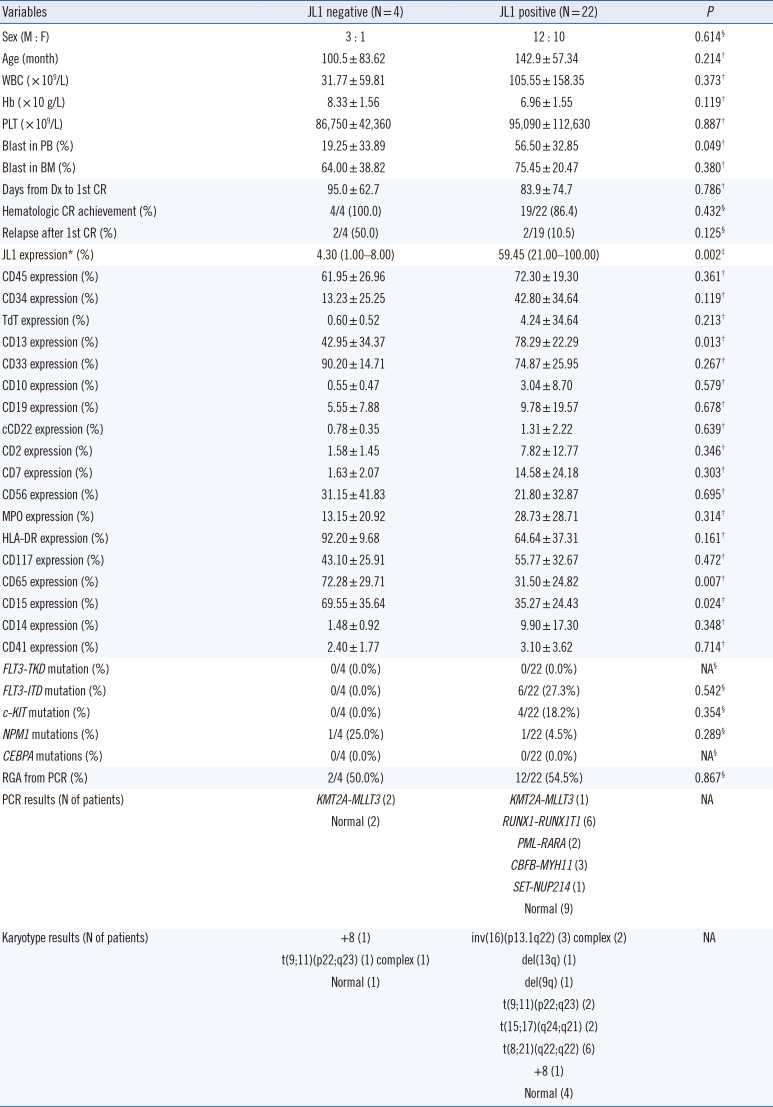
The clinical characteristics, including prognosis, of the 26 AML patients did not significantly differ with respect to the JL1 expression status. Patients with positive JL1 expression showed higher JL1 expression in leukemic blasts than those without JL1 expression (P=0.002). Notably, the patients with positive JL1 expression showed higher CD13 expression (P=0.013) and lower CD65 and CD15 expressions (P=0.007 and 0.024, respectively) in leukemic blasts than those without JL1 expression.
The patients with positive JL1 expression also showed a high incidence of RUNX1-RUNX1T1 (N=6), CBFB-MYH11 (N=3), and PML-RARA (N=2) rearrangements (detected by PCR), indicating the presence of t(8;21)(q22;q22), inv(16)(p13.1q22), and t(15;17)(q24;q21) chromosomal aberrations, respectively. The presence of these three chromosomal aberrations in 11 patients was further confirmed by karyotype analysis. These three genetic abnormalities were not observed in patients without JL1 expression.
Patients with positive JL1 expression showed an incidence of 27.3%, 18.2%, and 4.5% for the FLT3-ITD, c-KIT, and NPM1 mutations, respectively; patients without JL1 expression showed an NPM1 mutation incidence of 25.0%. However, the incidences of these three genetic mutations (FLT3-ITD, c-KIT, and NPM1) did not differ significantly with respect to the JL1 expression status (Table 3).
DISCUSSION
We found that the incidence of positive JL1 expression was 76.9% and 84.6% in de novo pediatric ALL and AML patients, respectively. Although these results are slightly lower than those reported previously (80.9–87.0% for ALL and 90.1% for AML) [7], we demonstrated that approximately 80% of de novo pediatric acute leukemia patients significantly express JL1 antigens in leukemic blasts. This finding may provide an adequate rationale for using the JL1 antigen as a candidate target for immunotherapy.
In addition, we found that ALL patients with positive JL1 expression have higher CD10 and cIgM expressions than those without JL1 expression. As the absolute value of cIgM expression was low, we hypothesize that the clinical significance of differences in cIgM expression is limited. However, the absolute value of CD10 expression in leukemic blasts of patients with positive JL1 expression was high, suggesting that pediatric patients with CD10-expressed common cell ALL are more likely to benefit from anti-JL1 immunotherapy than other patients. The heterogenous expression of the JL1 antigen on CD34+ and CD10+ lymphoid precursors was also reported in a previous study [11], which supports our results. In addition, we demonstrated that only de novo pediatric ALL patients with positive JL1 expression have TCF3-PBX1 and KMT2A-MLLT1 rearrangements, indicating the presence of t(1;19)(q23;p13.3) and t(11;19)(q23;p13.3) chromosomal abnormalities, respectively. Our results also suggest that anti-JL1 immunotherapy could be an additional therapeutic option for pediatric ALL patients who harbor TCF3-PBX1 and KMT2A-MLLT1 rearrangements, which result in a poor prognosis in infantile leukemia.
Interestingly, our results showed that all four T-ALL patients exhibited JL1 expression, which may indicate that the incidence of JL1 expression in pediatric T-ALL might be higher than in pediatric B-ALL. However, as the number of pediatric T-ALL patients included in our study was very limited, this result should be interpreted with much caution and should be further addressed in future studies.
Compared with ALL, the number of pediatric AML patients without JL1 expression was low, and the statistical power was limited. However, we demonstrated that de novo pediatric AML patients with positive JL1 expression have higher CD13 and lower CD65 and CD15 expressions than those without JL1 expression. These results suggest that pediatric AML patients with positive JL1 expression tend to present a more immature leukemic blast immunophenotype (lower expression of the granulocytic maturation markers CD65 and CD15) than those without JL1 expression.
We also showed that de novo pediatric AML patients with positive JL1 expression, but not those without JL1 expression, exclusively present with RUNX1-RUNX1T1, CBFB-MYH11, and PML-RARA rearrangements, which indicate the presence of t(8;21)(q22;q22), inv(16)(p13.1q22), and t(15;17)(q24;q21) chromosomal abnormalities. These results suggest that the application of anti-JL1 immunotherapy could be an additional therapeutic option for pediatric patients with core binding factor (CBF) AML and acute promyelocytic leukemia.
Moreover, although the differences were not statistically significant, we found that the incidence of the poor prognostic marker, FLT3-ITD and c-KIT mutations, is higher, and that of favorable prognostic markers, such as NPM1 mutations, is lower in de novo pediatric AML patients with positive JL1 expression than in those without JL1 expression. These results suggest that anti-JL1 immunotherapy could be an additional therapeutic option for pediatric AML patients who harbor poor molecular prognostic markers. Although a previous study suggested that a gelonin-conjugated anti-JL1 monoclonal antibody immunotoxin could be developed as a potential immunotherapeutic agent for treating various types of JL1-positive acute leukemias [10], the therapeutic effect of the JL1 monoclonal antibody has not been validated by well-designed clinical trials. Our study supports the application potential of the anti-JL1 monoclonal antibody, especially when conjugated with toxic substances, for the treatment of JL1-expressing pediatric acute leukemias. Well-designed clinical trials focused on this issue are required to further validate this proposed therapy option.
Our study has major limitations in terms of statistical power due to the small number of patients in each subgroup, and insufficient analysis of prognosis, including overall and event free survival, due to high hematologic CR rates and low relapse rates. All our suggestions should be confirmed with large-scale comprehensive studies, and the prognostic value of JL1 antigen expression needs to be investigated using well-controlled prospective studies.
In conclusion, we showed that 76.9% and 84.6% of de novo pediatric ALL and AML patients demonstrated positive JL1 expression, which supports the potential therapeutic role of anti-JL1 monoclonal antibodies. Positive JL1 expression cases were associated with specific immunophenotypic features and genetic abnormalities; however, the JL1 expression status did not significantly affect prognosis. This should be further examined in more comprehensive studies.

 XML Download
XML Download