

 PDF
PDF Citation
Citation Print
Print



Abbreviations
BCR
B cell antigen receptor
cGAS
cyclic GMP-AMP synthase
CTLD
C-type lectin like domain
FDC
follicular dendritic cell
GWAS
genome-wide association studies
ITIM
immunoreceptor tyrosine-based inhibition motif
NA
nucleic acid
PAMP
pathogen-associated molecular pattern
pDC
plasmacytoid dendritic cell
SHP-1
SH2-containing protein tyrosine phosphatase 1
SLE
systemic lupus erythematosus
INTRODUCTION
Systemic lupus erythematosus (SLE) is the prototype of systemic autoimmune disease characterized by production of autoantibodies to various nuclear self-antigens. Immune complexes composed of autoantibodies and self-antigens cause tissue damage including glomerulonephritis. Genetic studies on SLE patients and analyses of mice that develop lupus-like disease have demonstrated a crucial role of nucleic acid (NA) sensors, innate receptors that recognize NAs and activate immune cells, in development of SLE (12). NA sensors induce immune responses to microbes especially viruses by recognizing microbial NAs (345). NA sensors also recognize self-NAs and induce antibody production to NA-related nuclear self-antigens, which is crucial in development of SLE, and overexpression of genes regulated by IFN-I called IFN signature, the most prominent gene expression feature of SLE patients (678). CD72 is an inhibitory B cell co-receptor (9). Genetic analysis of both human SLE patients (10) and the mouse lupus model MRL-Faslpr/lpr (11), and analysis of CD72−/− mice (1213) show that CD72 prevents development of SLE. We previously demonstrated that CD72 recognizes an RNA-related lupus self-antigen Sm/RNP as a ligand, and negatively regulates B cell responses to this self-antigen (14). Thus, NA sensors and CD72 are activating and inhibitory receptors, respectively, capable of recognizing NA-related self-antigens. In this review, I will discuss the opposing roles of NA sensors and CD72 in the regulation of development of SLE.
THE ROLE OF NA SENSORS IN SLE
Immune cells express various NA sensors that transmit activation signaling upon recognition of NAs (34). NA sensors are involved in host defense against microbes especially viruses by recognizing microbial NAs. NA sensors are present in either endosome or cytoplasm. NA-recognizing TLRs such as TLR3, TLR7, TLR8, and TLR9 are present in endosome, whereas the NA sensors RIG-I, MDA5, and cyclic GMP-AMP synthase (cGAS) are located in cytoplasm.
Genome-wide association studies (GWAS) on SLE patients already determined more than 80 genetic loci associated with SLE (27). Although the contribution of each loci to the development of SLE is small, the list of the SLE-associated genes suggests the mechanisms for the development of SLE. This list includes genes encoding NA sensors such as TLR7 and IFIH1, the latter of which encodes MDA5, and those encoding molecules involved in signaling through NA sensors such as IRF5 and IRAK1. Genes encoding nucleases involved in NA degradation such as TREX1 and RNaseH2 are also associated with SLE. Defects in NA degradation may augment activation of NA sensors. These findings suggest that NA sensors play a role in development of SLE. The role of NA sensors in the development of SLE has also been suggested by studies on mouse models. Lupus-like disease is induced by a gain-of-function mutation of the NA sensor IFIH1 (15). Moreover, deficiency of the endosomal RNA sensor TLR7 completely inhibits development of lupus-like diseases in multiple different lupus models including MRL-Faslpr/lpr mice (16) and pristane-induced lupus (17). In contrast, the endosomal DNA sensor TLR9 rather reduces the disease severity (16) by competing transport of TLR7 to endosome (18). Thus, recognition of RNA-related nuclear self-antigens such as Sm/RNP but not DNA by NA sensors is crucial in development of SLE.
Recognition of self-NAs by NA sensors induces activation of B cells reactive to self-NAs (1920). Because B cell antigen receptor (BCR)-mediated endocytosis is a major endocytosis pathway in B cells, exogenous NAs including RNA-related self-antigens such as Sm/RNP from dead cells are preferentially endocytosed by B cells reactive to these self-antigens by BCR-mediated endocytosis, resulting in translocation of these self-antigens to endosome. Endocytosed NAs then stimulate NA sensors in endosomes, and activate self-reactive B cells by the combination of BCR signaling and signaling through NA sensors, leading to production of autoantibodies to self-NAs (Fig. 1). Activation of B cells reactive to NA-related self-antigens appears to involve exogenous but not endogenous NAs because specific activation of these self-reactive B cells relies on BCR-mediated endocytosis. Autoantibodies form immune complexes with self-antigens, and then cause tissue damage.
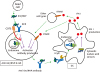 | Figure 1Immune response to NAs in development of SLE. The RNA-related lupus self-antigen Sm/RNP released from dead cells is recognized by BCR in Sm/RNP-reactive B cells, and generates BCR signaling. Sm/RNP is transported to endosome by BCR-mediated endocytosis, and stimulates the endosomal RNA sensor TLR7 essential in development of lupus thereby generating TLR7 signaling. Combination of BCR signaling and TLR7 signaling induces B cell activation and production of anti-Sm/RNP antibody. The immune complex consisting of Sm/RNP and anti-Sm/RNP antibody is endocytosed by DCs through interaction with Fcγ receptor, and is recognized by TLR7 in endosome, resulting in production of IFN-I. IFN-I is also produced through recognition of NAs by cytosolic NA sensors. IFN-I activates B cells directly through IFNAR on B cells or indirectly through activation of other cell types, thereby augmenting activation of self-reactive B cells. The inhibitory B cell co-receptor CD72 recognizes Sm/RNP, and specifically inhibits BCR signaling when BCR recognizes Sm/RNP, thereby inhibiting production of anti-Sm/RNP antibody. Originally published in F1000Research, Tsubata (2017) with modification (8).
|
NA sensors also contribute to development of SLE by inducing production of IFN-I. Analysis of peripheral blood cells from SLE patients show augmented expression of IFN-I-regulated genes (IFN signature) (2122). Plasmacytoid dendritic cells (pDCs) are thought to be a major IFN-I producer in SLE because pDCs are capable of producing a large amount of IFN-I (23). Immune complexes composed of NA-related self-antigens and autoantibodies are endocytosed by pDCs through FcγR-mediated endocytosis, and induces IFN-I production by stimulating TLRs in endosome (24). IFN-I enhances activation of B cells either directly or indirectly thereby augmenting autoantibody production (25). Thus, autoantibodies from B cells and IFN-I from pDCs form a positive feedback loop, resulting in production of large amounts of both autoantibodies and IFN-I. Although pDCs are thought to be a major IFN-I producer, other cell types such as follicular dendritic cells are also shown to play a role in IFN-I production and contribute to development of SLE (26).
NA-induced IFN-I production involves cytosolic NA sensors such as cGAS, MDA5 and RIG-I (7) as well as endosomal TLRs. Mutations of enzymes involved in NA metabolism such as TREX1, RNaseH2, SAMHD1, and ADAR1 cause dysregulated IFN-I production through activation of cytosolic NA sensors by NAs accumulated in cytosol. These mutations cause interferonopathies including Aicardi-Goutier syndrome, in which augmented IFN-I production induces systemic inflammatory disease (27). Various rare mutations in TREX1 are found in 1%–2% of SLE patients (2829), suggesting a role of TREX1 mutation in development of SLE in these patients. TREX1 prefers single-stranded DNA (3031) and the nicked strand of double-stranded DNA (3233) as substrates, and is suggested to catalyze cDNA derived from retroelements because reverse transcriptase inhibitors ameliorate inflammatory disease in TREX1−/− mice (34). The disease in TREX1−/− mice depends on cGAS (35). Thus, accumulated endogenous NAs such as cDNAs of retroelements may induce IFN-I production by activating cGAS in SLE patients with TREX1 mutation. Although cGAS senses cytosolic NAs, cGAS is activated by deficiency of the endosomal nuclease DNase II (3536), and is also required for IFN-I production induced by endocytosis of apoptotic bodies in SLE sera (37). Thus, cGAS may contribute to IFN-I production in SLE by sensing endocytosed NAs as well as endogenous cytosolic NAs.
Both B cells and pDCs (and other IFN-I-producing cells) appear to be involved in development of SLE by forming a positive feedback loop. In MRL-Faslpr/lpr mice, B cell-specific deletion of the adaptor molecule MyD88 required for TLR7 signaling strongly suppresses both autoantibody production and development of lupus nephritis (38). In contrast, autoantibody production is modestly reduced and nephritis is not suppressed by DC-specific deletion of MyD88. This result suggests that B cell response to NAs plays a central role in development of lupus. This notion is supported by the genome analysis of SLE patients. Genetic loci associated with SLE are enriched in genes specifically expressed in B cells but not other cell types including DCs (39). SLE-associated variations are also accumulated in super-enhancers, clusters of transcriptional enhancers, that play key roles in B cell biology (40). Based on the findings indicating crucial role of TLR7 and B cells in SLE, the response of endosomal RNA sensor TLR7 in B cells appears to be essential in development of lupus. TLR7 may activate B cells reactive to RNA-related self-antigens upon interaction with exogenous self-antigens from dead cells, thereby playing an essential role in development of lupus (Fig. 1).
CD72 IS AN INHIBITORY RECEPTOR THAT SPECIFICALLY SUPPRESSES DEVELOPMENT OF SLE
CD72 is a type 2 membrane protein of around 45 kDa mostly expressed in B cells. CD72 contains a C-type lectin like domain (CTLD) in the extracellular region and an immunoreceptor tyrosine-based inhibition motif (ITIM) in the cytoplasmic region. Upon phosphorylation, CD72 ITIM recruits and activates SH2-containing protein tyrosine phosphatase 1 (SHP-1) (41), which then negatively regulates signaling through BCR by dephosphorylating BCR proximal signaling molecules such as Syk (4243).
Involvement of CD72 in the regulation of lupus was first suggested by a genetic study of MRL-Faslpr/lpr mice (11). MRL-Faslpr/lpr mice develop severe lupus-like disease: Almost all female MRL-Faslpr/lpr mice produce large amounts of autoantibodies to nuclear self-antigens and develop immune complex nephritis by 6 months of age. Development of severe lupus-like disease requires Faslpr, which is a loss-of-function mutation of Fas (CD95) (44). However, Faslpr alone is not sufficient for development of severe lupus because C57BL/6 and C3H mice carrying Faslpr develop only a marginal disease (45). Thus, the MRL background contributes to the severe disease in MRL-Faslpr/lpr mice. A genetic analysis using MRL-Faslpr/lpr mice showed a genetic locus of MRL containing CD72 is associated with nephritis (11). There are three CD72 allelic forms, i.e., CD72a, CD72b and CD72c, in laboratory mice. MRL carries CD72c, whereas most of the other laboratory mouse strains such as C57BL/6 and BALB/c carry either CD72a or CD72b. CD72c contains a number of amino acid substitutions and a 7 amino acid deletion compared to CD72a or CD72b whereas CD72a and CD72b are highly conserved in amino acid sequence (46), suggesting a functional alteration in CD72c carried by MRL mice. Indeed, replacement of the genetic locus containing CD72c by that of CD72b derived from C57BL/6 mice significantly reduces disease severity in MRL-Faslpr/lpr mice (13). Conversely, C57BL/6-Faslpr/lpr mice carrying CD72c (C57BL/6.CD72c-Faslpr/lpr) develop a moderate disease whereas C57BL/6-Faslpr/lpr mice, which carry CD72b, develop only a modest disease at the 12 months of age (Fig. 2). Although C57BL/6.CD72c-Faslpr/lpr mice develop moderate lupus-like disease, MRL-Faslpr/lpr mice show more severe disease, indicating that the MRL background carries other genetic loci than CD72c that enhance the severity of the disease. Nonetheless, these findings clearly show that CD72 is associated with SLE and regulates development of SLE in MRL-Faslpr/lpr mice, and suggest that CD72c carried by MRL is a hypomorphic allele.
 | Figure 2CD72 regulates development of lupus-like disease in mice. (A-C) CD72c, a hypomorphic allele of CD72, causes moderate lupus-like disease on C57BL/6 (B6) background in the presence of the Faslpr mutation. Representative PAS staining (A) and immunohistochemistry for IgG and C3 (B) of kidney sections, and scores of the severity of glomerulonephritis (C) are shown. Originally published in The Journal of Immunology, Xu et al. (2013). CD72c is a modifier gene that regulates Faslpr-induced autoimmune disease. J Immunol. 190::5436-5445. Copyright © [2013] The American Association of Immunologists, Inc. (13). (D) CD72 activity inversely correlates to the severity of lupus-like disease. CD72c is a hypomorphic allele of CD72. Severity of the lupus-like disease in C57BL/6 (B6)-Faslpr/lpr and MRL-Faslpr/lpr mice inversely correlates to the activity of CD72 to inhibit BCR signaling. More severe disease in the mice on the MRL background carrying CD72c than those on the B6 background suggests that the MRL background contains (an) additional SLE-causing gene(s) other than CD72c. Originally published in Proceedings of the Japan Academy. Series B, Physical and Biological Sciences, Tsubata (2018) with modification (47).
|
Candidate gene approach revealed that an intronic polymorphism in the CD72 locus is associated with lupus nephritis in human (10). However, GWAS on SLE patients so far failed to show any association of CD72 with SLE. This is probably because there are no common variants of CD72 with a functional difference strong enough to show the association with SLE by GWAS. In future, GWAS and sequencing analyses with a larger number of patients might show association of CD72 with SLE or presence of rare variants of CD72 in SLE.
Regulation of the development of SLE by CD72 was further supported by observations on CD72−/− mice. Parnes and her colleagues first demonstrated development of lupus-like disease in CD72−/− mice (12). Our group analyzed CD72−/− mice more extensively, and demonstrated that CD72−/− C57BL/6 mice spontaneously develop relatively severe lupus like disease (13). Almost all these mice show glomerulonephritis at the age of 6 months. CD72−/− C57BL/6-Faslpr/lpr mice show more severe disease comparable to that in MRL-Faslpr/lpr mice (Fig. 2D). These results clearly indicate that CD72 prevents development of SLE.
B lymphocytes express various other inhibitory co-receptors such as CD22 (also known as Siglec-2), Siglec-10 (Siglec-G in mice), PECAM-1 (also known as CD31), LILRB (PIR-B in mice) and FcγRIIB (9). As is the case for CD72, these inhibitory co-receptors contain ITIMs in the cytoplasmic region. Upon phosphorylation, these ITIMs recruit and activate SH2-containing phosphatases such as SHP-1 and SHIP-1. Most of the inhibitory co-receptors including CD22, CD72, Siglec-10/G, PECAM-1 and LILRB/PIR-B activate SHP-1, which inhibits B cell activation by negatively regulating BCR signaling (Fig. 3).
 | Figure 3Differential functional properties of inhibitory B cell co-receptors. B cells express various inhibitory B cell co-receptors such as CD22 (also known as Siglec-2), CD72, Siglec-10 (Siglec-G in mouse), LILRB (PIR-B in mouse), PEACAM1 (also known as CD31), PD-1 and FcγRIIB. These receptors contain ITIMs in the cytoplasmic region and recruit SH2-containing phosphatases such as SHP-1, SHP-2, and SHIP-1 upon phosphorylation by the BCR-associated kinase Lyn, leading to down-modulation of BCR signaling. Although many of these inhibitory receptors activate SHP-1, CD72, and Siglec-G inhibit development of lupus-like disease and B-1 cell expansion, respectively, and CD22 regulates development and phenotype of conventional B cells. Originally published in Frontiers in Immunology, Tsubata (2018) with modification (9).
|
B cell-specific deletion of SHP-1 causes diverse phenotypes such as 1) severe lupus-like autoimmune disease, 2) expansion of B-1 cells, and 3) alterations in the development and phenotype of conventional B cells (48). In contrast, deletion of each SHP-1-activating inhibitory B cell co-receptor does not show all these diverse phenotypes, but shows only a restricted phenotype depending on the inhibitory co-receptor. As a consequence, deletion of an inhibitory B cell co-receptor causes a distinct phenotype (Fig. 3). CD22−/− mice show alterations of conventional B cells similar to SHP-1−/− conventional B cells (4849505152), whereas Siglec-G−/− mice show marked expansion of B-1 cells (53). CD72−/− mice show lupus-like disease but not alterations in development and phenotype of conventional B cells or B-1 cell expansion (13). Thus, although SHP-1 regulates various distinct B cell populations including B-1 cells, conventional B cells and self-reactive B cells, each SHP-1-activating inhibitory B cell co-receptor regulates distinct B cell population and phenotypes. Mice deficient in inhibitory B cell co-receptors except for CD72−/− mice develop either modest or no lupus-like disease. Indeed, CD22−/− mice and PIR-B−/− mice do not develop lupus-like disease (5455). PECAM1−/− mice and Siglec-G−/− mice develop lupus-like nephritis only in a fraction of mice even at 12 months of age (5456). Because almost all CD72−/− mice develop lupus-like nephritis already at 6 months of age, CD72 is the major regulator of lupus-like disease among inhibitory B cell co-receptors.
CD72 REGULATES B CELL RESPONSES TO THE RNA-RELATED SELF-ANTIGEN Sm/RNP
Although most of the inhibitory B cell co-receptors regulate B cell activation by recruiting SHP-1, each SHP-1-activating inhibitory B cell co-receptor recognizes distinct ligands by a distinct ectodomain (9). The ligand of CD72 was initially reported to be CD5, although this result has not been reproduced (57). Later, CD100 (also known as Semaphorin-4D) was shown to be an inhibitory ligand of CD72 (58). The functional significance of this inhibitory ligand is not yet clear. We thus determined the ligand of CD72 to address why CD72 specifically regulates development of lupus-like disease. We demonstrated that a recombinant CD72 CTLD specifically binds to Sm/RNP (14), suggesting that CD72 recognizes Sm/RNP as a ligand. Moreover, CD72 is phosphorylated at ITIM, recruits SHP-1, and inhibits BCR signaling when Sm/RNP stimulates B cells as an antigen and ligates BCR. In contrast, CD72 is not phosphorylated or inhibits BCR signaling when BCR is ligated by control antigens. Thus, recognition of Sm/RNP as a ligand appears to induce CD72 phosphorylation and CD72-mediated signal inhibition when BCR is ligated by Sm/RNP. The scenario how CD72 specifically regulates Sm/RNP-induced BCR signaling is as follows. When BCR is ligated by Sm/RNP, CD72 is recruited to the close vicinity of BCR by interacting BCR-bound Sm/RNP, resulting in phosphorylation of CD72 ITIM by the BCR-associated kinases Lyn (Fig. 4). The phosphorylated ITIM activates SHP-1 and induces SHP-1-mediated inhibition of BCR signaling. Other antigens do not induce CD72-mediated signal inhibition in the absence of interaction of CD72 with the BCR-bound antigens. As already discussed earlier in this article, B cell responses to RNA-related self-antigens such as Sm/RNP plays a crucial role in the development of SLE. By inhibiting Sm/RNP-induced BCR signaling, CD72 appears to induce B cell tolerance to Sm/RNP and thereby prevent development of SLE (Fig. 1). CD72c shows weaker binding to Sm/RNP compared to CD72a (14). This finding supports the notion that CD72c is a hypomorphic allele. CD72c may not be able to inhibit BCR signaling induced by Sm/RNP strongly enough to completely prevent activation of self-reactive B cells, thereby contributing to development of severe lupus-like disease in MRL-Faslpr/lpr mice. This was the first demonstration of the mechanism in which an inhibitory receptor prevents pathogenic immune responses to self-antigens by recognizing the self-antigens, and prevents development of autoimmune disease.
 | Figure 4CD72 specifically inhibits BCR signaling in Sm/RNP-reactive B cells by recognizing Sm/RNP as a ligand. When Sm/RNP binds to Sm/RNP-reactive BCR, CD72 is recruited to the close proximity of BCR by binding to Sm/RNP bound to BCR (A). CD72 ITIM is then phosphorylated by the BCR-associated kinase Lyn and recruits SHP-1. This causes suppression of BCR signaling in Sm/RNP-reactive B cells thereby inhibiting production of anti-Sm/RNP antibody crucial for development of lupus. When other antigens that do not bind to CD72 interact BCR, CD72 is kept away from BCR and does not inhibit BCR signaling (B). Originally published in Frontiers in Immunology, Tsubata (2018) (9).
|
Does ligand recognition of other inhibitory B cell co-receptors than CD72 regulate their functional properties? Both Siglec-G and CD22 are members of sialic acid-binding immunoglobulin-like lectin (Siglec) family. Both of them recognize sialic acid as a ligand. However, Siglec-G recognizes both α2,3 and α2,6 sialic acid whereas CD22 specifically recognizes α2,6 sialic acid (5960). Nitschke and his colleagues demonstrated that B-1 cells express α2,3 sialic acid much more abundantly than conventional B cells. They also showed that Siglec-G interacts with BCR on the same cell by recognizing sialic acid on BCR (61). These findings suggest that Siglec-G but not CD22 is recruited to the close vicinity of BCR that are dominantly α2,3-sialylated in B-1 cells, and phosphorylated at ITIM by the BCR-associated kinases, leading to signal inhibition in B-1 cells. Thus, ligand binding of CD72 and Siglec-G determines which BCR is efficiently regulated by these inhibitory co-receptors, i.e., Sm-RNP-reactive BCR by CD72 and α2,3-sialylated BCR expressed in B-1 cells by Siglec-G. Regulation of distinct BCR by CD72 and Siglec-G determines the B cell function regulated by these inhibitory co-receptors: CD72 regulates development of lupus-like disease whereas Siglec-G regulates B-1 cells. Thus, distinct ligand recognition plays a crucial role in determining the distinct functional properties of inhibitory B cell co-receptors (Fig. 3).
Microbial NAs are recognized by various NA sensors as one of the major pathogen-associated molecular patterns that play a role in induction of immune responses against microbes. However, self NAs must be discriminated from microbial NAs to avoid autoimmunity. NA sensors including TLR7 respond to microbial NAs better than endogenous NAs by recognizing the structural features of microbial NAs such as dsRNA and 5′-triphosphate RNA (62). Intracellular localization of NA sensors may also contribute to discrimination of self vs. microbial NAs. Self NAs from dead cells may be readily digested by nucleases in the body fluid whereas microbial NAs are exposed after introduction to the host cells. The importance of the localization of NA sensors in maintenance of self-tolerance is supported by the observation that expression of the endosomal NA sensor TLR9 on the cell surface induces autoimmunity (63). Besides these mechanisms, CD72 contributes to the discrimination of microbial NAs from self NAs by suppressing B cell response to nuclear self-antigens containing self-RNA.
CONCLUSIONS
Multiple lines of evidence strongly suggest that NA sensors play a central role in development of SLE. Although NA sensors induce anti-microbial immune responses by recognizing microbial NAs, NA sensors also recognize self NAs. Immune responses to self NAs mediated by NA sensors induce production of autoantibodies to nuclear self-antigens from B cells and production of IFN-I from pDCs, both of which form a positive feedback loop by enhancing each other, thereby contributing to the development of SLE. Among NA sensors, the endosomal RNA sensor TLR7 is particularly important at least in mouse lupus models (1617). In contrast, the inhibitory B cell co-receptor CD72 recognizes the RNA-containing endogenous TLR7 ligand Sm/RNP, and inhibits B cell response to Sm/RNP (14). By suppressing TLR7-dependent B cell responses to RNA-related self-antigens, CD72 contributes to discrimination of self-NAs from microbial NAs, and prevents development of lupus.
The anti-malaria drug hydroxychloroquine has been used for the treatment of SLE since more than 50 years ago (64). Because of its beneficial effects on SLE, hydroxychloroquine is now recommended to use for treatment of SLE regardless of the severity of the disease (65). The mechanism how hydroxychloroquine controls SLE has been demonstrated since around 15 years ago (6667) after discovery of the role of NA sensors in development of lupus. Hydroxychloroquine inhibits activation of endosomal NA sensors including TLR7 probably by preventing acidification of endosome, which is required for optimal NA recognition by endosomal NA sensors. This finding suggests that the NA sensors are good target molecules for development of more effective drugs for SLE. Further development of new drugs targeting NA sensors and also CD72 may achieve better control of SLE.
 XML Download
XML Download