

 PDF
PDF Citation
Citation Print
Print



Abbreviations
ACPA
anti-citrullinated peptide antibody
ARTD1
ADP-ribosyltransferase diphtheria toxin-like 1
BMM
bone marrow macrophage
DAMP
damage-associated molecular pattern
FLS
fibroblast-like synoviocytes
ITAM
immunoreceptor tyrosine-based activation motif
MCV
mutated citrullinated vimentin
MSU
monosodium urate
NET
neutrophil extracellular trap
NLRP3
NOD leucine rich repeat with a pyrin domain 3
NOD
nucleotide oligomerization domain
OC
osteoclast
OSCAR
osteoclast-associated receptor
PAD
peptidylarginine deiminase
PARP1
poly(ADP-ribose) polymerase 1
RA
rheumatoid arthritis
RANKL
RANK ligand
RBP-J
recombination signal binding protein for immunoglobulin kappa J region
SLE
systemic lupus erythematosus
TRAF6
TNF receptor-associated factor 6
TREM
triggering receptor expressed on myeloid cells
INTRODUCTION
Bone erosion and joint destruction is a characteristic finding of some inflammatory arthritis including rheumatoid arthritis (RA), psoriatic arthritis, and gout, and is associated with functional disability and increased mortality (12). The activation of osteoclasts (OCs), which is differentiated from myeloid OC precursors of the monocyte-macrophage lineage, is responsible for bone erosion in inflammatory arthritis (3). The origin of OCs in RA which is mainly located at the junction of synovial pannus and bone is not clear until now. Immature dendritic cells being rich in inflamed synovium can directly transdifferentiated into OCs in the inflammatory condition of RA (4). The presence of tartrate-resistant acid phosphatase-positive OCs on both synovial and marrow sides of subchondral bone suggests that subchondral bone marrow also can be the origin of OCs in RA (5). The treatment goal for bone-erosive inflammatory arthritis is to minimize the structural joint damage as well as suppress inflammation itself (6). To achieve this goal, the mechanisms for differentiation and activation of OCs under inflammatory milieu have been actively investigated and its therapeutic application to inhibit the functions of OCs has been in the limelight. In this review, we will address the scientific achievements about the molecular mechanisms involving OC differentiation under an inflammatory condition, especially in RA.
ENHANCED EXPRESSION OF RANK LIGAND (RANKL) AND OSTEOCLAST-ASSOCIATED RECEPTOR (OSCAR) IN INFLAMMATORY ARTHRITIS
Interaction between the RANK and RANKL is critical for triggering OC precursors to differentiate into OCs (7). RANKL binds to RANK on the surface of OC precursors results in the recruitment of the adaptor molecule, TNF receptor-associated factor 6 (TRAF6), which activates NF-κB, AP-1 (c-Fos and c-Jun), MAP kinases, and phospholipase Cγ (7) (Fig. 1A). RANKL is mainly released by osteoblast and osteocyte during the process of physiologic bone remodeling (89). However, this role can be replaced by immune cells and fibroblast-like synoviocytes (FLS) in RA condition (10111213). Synovial B cells from RA patients are enriched with switched memory B cells (CD27+IgD−) and spontaneously express RANKL which is much higher than that of synovial T cells (10). Actually, B cell depletion therapy with rituximab significantly abrogates the joint erosion, which is associated with a decrease of synovial OC precursors and RANKL expression (1415). In terms of T cells, Th17 cells express a significant amount of RANKL, but Th1 and Th2 cells express only a minimal amount (1112). FLS can also be the major source of RANKL which is induced by inflammatory cytokines such as TNFα, IL-17, and IL-6 in RA (13). Recent study using conditional deletion of RANKL in T cells or FLS revealed that RANKL of FLS has a primary role compared to RANKL of T cells in bone erosion of inflammatory arthritis (16).
Figure 1
Inflammatory cytokines such as TNFα, IL-1β, and IL-6 can replace the function of RANKL/RANK signaling during OC differentiation. (A) Schematic signaling pathways being critical for OC differentiation in physiologic condition. RANKL/RANK signaling activates NF-κB and c-Fos through TRAF6, leading to the transcription of NFATc1. Calcium signaling through ITAM motif of FcRγ or DAP12 increases nuclear translocation of NFATc1 through its dephosphorylation, and NFATc1 in turn promotes its own transcription within the nucleus, forming auto-amplification loop. (B) TNFα together with IL-1β or IL-6 can substitute for RANKL/RANK signaling through the activation of NFkB, c-Fos and NFATc1 which are essential transcription factors for OC differentiation.
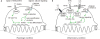
Together with RANKL-RANK signaling, the immunoglobulin-like receptor, OSCAR and triggering receptor expressed on myeloid cells (TREM)-2 that is associated with the immunoreceptor tyrosine-based activation motif (ITAM)-containing FcRγ and DAP12, respectively, leads to the activation of calcium signaling, which induces the auto-amplification of the NFATc1, master transcription factor for osteoclast differentiation (1718) (Fig. 1A). Compared to controls, peripheral blood monocytes from RA patients express a higher level of OSCAR which is mainly induced by TNFα and associated with disease activity of RA (19). Actually, synovial tissues from active RA patients express higher levels of OSCAR than OA and control. OSCAR is mainly expressed by OCs at the erosion and by mononuclear cells around synovial microvessels (1920). Ligands for OSCAR is mainly type I (ColI) and type II collagen (ColII) which are the most abundant collagen of bone and cartilage, respectively (21). Among collagens, ColII peptides efficiently stimulate RANKL-dependent OC differentiation (21). In addition to OCs, ITAM signaling from OSCAR-ColI/II interaction promotes survival and cytokine production of monocytes (22), and dendritic cells (23). TREM-2 is also highly expressed in the synovial tissue of RA, but it is not well elucidated the pathologic role in OC differentiation of RA (20).
INFLAMMATORY CYTOKINES AND OC DIFFERENTIATION: INTERACTION BETWEEN TNFα, IL-6, AND IL-1
RA is a prototype of chronic autoimmune arthritis with an increase of inflammatory cytokines including TNFα, IL-6, and IL-1 which can also affect the OC differentiation (24). TNFα can dramatically enhance OC differentiation in the presence of low level of RANKL that is insufficient to induce OC formation, while TNFα alone without RANKL failed to induce the differentiation of OCs (25). However, TNFα can induce OC differentiation in the recombination signal binding protein for immunoglobulin kappa J region (RBP-J)-deficient cells even in the absence of RANKL, suggesting RBP-J, a key mediator of signaling by the canonical Notch pathway, acts as upstream negative regulator of TNFα-mediated OC differentiation (26). In addition, the higher dose of TNFα (20 ng/ml) can induce OC differentiation without RANKL, but it fails for OCs to have a resorbing capacity of dentin slice (27).
Considering RANKL is a member of TNF superfamily, it is not surprising the induction or enhancement of OC differentiation by TNFα sharing signaling pathways with RANKL (2829). However, TNFα alone cannot produce complete OCs with resorption function (27). These limits of TNFα in OC differentiation are overcome by the presence of other inflammatory cytokines such as IL-1 and IL-6 (2730). IL-1 and IL-6 can promote OC differentiation in the presence of RANKL in vitro, but they cannot differentiate OC on its own without RANKL (3132). However, IL-1 together with TNFα can differentiate mouse bone marrow macrophages (BMMs) into functional OCs (27). IL-6 in the presence of TNFα also generates functional OCs in vitro which is independent to RANK/RANKL signaling (30). This TNFα and IL-6-mediated OC differentiation does not occur in the BMMs from NFATc1 or DAP12-defective mice (30), meaning that the differentiation into OC is possible regardless of ligand and receptor specificity when NFATc1 is induced by NF-κB and AP-1 (Jun/Fos complex) signaling, and is auto-amplified by the calcium signaling (Fig. 1B).
T-CELL-MEDIATED REGULATION OF OC DIFFERENTIATION
Bone erosion of the involved joints is a characteristic finding in RA, but it rarely occur in the arthritis of systemic lupus erythematosus (SLE), even in the 5%–15% of patients with long-standing lupus arthritis who develop deformities by a subluxation of ligaments, known as Jaccoud's arthropathy (33). The synovial inflammation of RA is mainly driven by M1 macrophages and Th17 cells, and the main pathogenic mechanism of SLE is humoral immunity characterized by autoantibodies against nuclear and cytoplasmic antigens (3435). This suggests that even if there is synovitis in both RA and SLE, the development of bone erosions depends on the context of inflammatory milieu determined by T cell subsets and their cytokines.
INFγ, the main Th1 cytokine, strongly suppresses OC differentiation in vitro through the proteosomal degradation of TRAF6 (36). It also downregulates RANKL-mediated cathepsin K expression in OC precursors which is critical for both differentiation and function of OCs (37). IL-4 as a Th2 cytokine is known to suppress OC differentiation through PPARγ and STAT6 activation (3839). On the other hand, the co-culture with Th17 cells enhances OC differentiation through not only the action of IL-17, but also RANKL expression (11). Th17 cytokines including IL-17, IL-21, and IL-22 is mainly responsible for the bone erosion in RA through direct induction of OC differentiation as well as RANKL production from FLS and osteoblast (114041). The blocking antibody against IL-17A inhibits OC differentiation in vivo, which is associated with the induction of IL-12 and IL-4, and the increase of Th2 and Tregs (42). Another T cell subset, Tregs, suppresses OC formation. Co-culture of Tregs suppresses OC differentiation from OC precursors as well as their bone resorbing function in vitro (43). The transgenic mice of Foxp3 that is the master regulator of Tregs revealed an osteopetrotic phenotype by the suppression of OC (44). Treg-mediated inhibition of OC differentiation is largely dependent on direct cell-cell contact via the CTLA-4, whereas TGFβ and IL-10, the major cytokines of Tregs, did not have an essential role (43). Abatacept that is a fusion protein with the extracellular domain of CTLA-4 inhibited OC formation in a dose-dependent manner in vitro, and successfully attenuated the bone erosion in the arthritis of TNF transgenic mice (45). Taken together, these evidences suggest that the distribution of T cell subsets that is different according to inflammatory diseases can influence the cytokine milieu, and the net effects of T cell subsets and their cytokines can determine the differentiation of OC and consequently the erosive phenotype of the individual inflammatory diseases (Fig. 2).
Figure 2
Osteoclastogenesis is carefully orchestrated by various cytokines and immune cells in RA environment. FLS produces RANKL and TNFα which is induced by inflammation milieu, and macrophages are the main source of inflammatory cytokines such as TNFα, IL-1β, and IL-6 which can induce OC differentiation. B cells mainly express RANKL in RA synovium. In terms of T cell subset, Th17 cells increase OC differentiation through RANKL and IL-17 production, while Treg, Th1, and Th2 cells suppress it.

OC DIFFERENTIATION BY ANTI-CITRULLINATED PEPTIDE ANTIBODY (ACPA)
Citrullination is the post-translational modification of the amino acid arginine with positive charge at a neutral pH into the citrulline with neutral charge by peptidylarginine deiminase (PAD) enzyme in a calcium-dependent manner (4647). The citrullinated target proteins such as filaggrin and vimentin lose their positive charge that was maintained by arginine, and consequently induce conformational changes of proteins (47). Arginine is essentially incapable of anchoring within the first binding pocket of all HLA-DR proteins because of its charge and relatively large size (48). Whereas the citrullinated peptides can have preferential but weak binding to HLA-DR proteins and it can make the citrullinated self-antigen-specific T cells escape the thymic negative selection (4950). These CD4+ T cells actively contribute to the production of ACPA which is highly specific for the RA (4647). Mounting evidences suggest the mucosal site such as lung and gingiva can be responsible for the initial ACPA production which is associated with long-term exposure to cigarette smoking and chronic periodontitis by Porphyromonas gingivalis (5152).
RA is chronic inflammatory disorder characterized by periarticular bone erosion that is associated with disease severity and poor functional outcome (53). Recent evidences found that ACPA is involved in the development of RA as well as bone erosion through OC differentiation (5455). Even the subjects with ACPA who have no clinical symptom of RA, namely preclinical RA, showed a reduced bone mineral density which was mainly by cortical bone thinning and porosity, and a higher incidence of erosions in metacarpophalangeal joints compared to ACPA-negative controls (56). This result suggests that ACPA alone can trigger OC activation even in the absence of active inflammation. OCs and OC precursors express not only vimentin in their cytoplasm, but also PAD2 and PAD4 enzymes, which is unique for OCs and OC precursors, but not other cells in the joint tissue (555758). Treatment of ACPA against mutated citrullinated vimentin (MCV) not only bound to osteoclast surfaces, but also led to robust induction of in vitro OC differentiation and bone-resorptive activity (54). This enhanced OC differentiation was reproduced in adoptive transfer model of MCV-ACPA in vivo, resulting in osteopenic phenotype (54). ACPA-mediated enhancement of OC differentiation was blocked by PAD enzyme inhibitor in a dose-dependent manner (55). Collectively, these evidences suggest that the vimentin is citrullinated by PAD enzyme in OCs and OC precursors, and this citrullinated vimentin expressed on the cell surface allows ACPA to bind to OC, which promotes OC differentiation and activation (Fig. 3).
Figure 3
ACPA with specificity to citrullinated vimentin induces the differentiation of OC precursors as well as promotes the inflammatory response in RA. The vimentin located in the cytoplasm of OC precursors and OCs is citrullinated by PDA enzymes in the presence of calcium and it is expressed on the cell surface. ACPA, which appears to have been generated in the inflammatory environment of lung or gingiva, binds to this citrullinated vimentin and it increases the expression of IL-8 from OC precursors and OCs. IL-8 increases the differentiation of OC precursors in an autocrine manner and is also involved in the recruitment of neutrophils as a chemokine. NETs formation by recruited neutrophils acts to amplify the inflammation of synovial tissue and thus to develop RA.
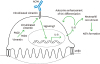
Although the precise signaling mechanisms are not well known, the binding of ACPA to the citrullinated vimentin on OC surface triggers the production of IL-8, also known as CXCL8 (5558). ACPA-mediated OC differentiation is completely abolished by IL-8 neutralization or its chemical inhibitor, suggesting that IL-8 acts as an autocrine growth factor for OCs in the presence of ACPA (55). In addition, IL-8 as a potent chemokine can recruit neutrophils to the joint tissue. These activated neutrophils release neutrophil extracellular traps (NETs) consisted of processed chromatin bound to granular and selected cytoplasmic proteins (59). NETs can not only be a huge source of citrullinated antoantigen, but also stimulate inflammatory response through cytokine release, and activation of FLS and macrophage in RA (60).
Collectively, the osteoclast differentiation normally controlled by osteoblast- or osteocyte-secreted RANKL can be amplified under inflammatory condition of RA. The osteoclast-mediated bone destruction in RA is finely regulated not only by immune cells such as T cells, B cells and macrophages, and their cytokines, but also by FLS and RA-specific antibody of ACPA.
INNATE IMMUNE SYSTEM AND OC DIFFERENTIATION
Gout, along with RA, is another inflammatory arthritis characterized by bone erosion, which is caused by precipitation of monosodium urate (MSU) crystal in the joint (61). MSU crystal as a damage-associated molecular patterns (DAMPs) triggers innate immune response through pattern recognition receptors, such as nucleotide oligomerization domain (NOD)-like receptors and Toll-like receptors (TLRs), which implicates a role of innate immune system in OC differentiation (6162).
MSU crystals phagocytosed by macrophages are recognized by the NOD leucine rich repeat with a pyrin domain 3 (NLRP3) inflammasome involving the activation of procaspase-1, which in turn cleaves proIL-1β to active IL-1β (61). Actually, MSU-mediated enhancement of OC differentiation is significantly attenuated by the knockdown of IL-1β (63). NLRP3-induced activation of procaspase-1 can also trigger the cleavage of ADP-ribosyltransferase diphtheria toxin-like 1 (ARTD1), also known as poly(ADP-ribose) polymerase 1 (PARP1), into 89 kDa- and 24 kDa-sized fragments, leading to the loss of its enzyme function (64). This inhibition of ARTD1 results in the activation of canonical NF-κB signaling, which enhances the expression of IL-1β and NFATc1 in OC precursors (65). In addition, the OC-specific gain-of-function mutation of NLRP3 increases osteolysis in vivo resulting in 50% lower bone mass without systemic inflammation compared to control mice which is responsible for the enhanced reorganization of actin cytoskeleton (66). This result suggests a direct role of NLRP3 inflammasome in the function of mature OCs (Fig. 4).
Figure 4
DAMPs-NLRP3 inflammasome in OC differentiation and function. DAMPs such as MSU are endocytosed into OC precursors where TLRs may have a role. Upon exposure to DAMP, NLRP3 inflammasome is activated and in turn catalyses the conversion of procaspase-1 to caspase-1, which contributes to the production and secretion of the mature IL-1β. The activated caspase-1 also breaks down ARTD1, also known as PARP1, which leads to loss of its inhibitory role against canonical NF-κB signaling. The enhanced NF-κB activity can increase the transcription of IL-1β and NFATc1 that potentiate OC differentiation from OC precursors. In mature OC, NLPR3 inflammasome enhances bone resorbing ability through reorganization of actin cytoskeleton.
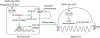
OC precursors also express TLRs, especially TLR2 and TLR4 (67). Although the ligands for TLR2 and TLR4 induces NF-κB activation and up-regulates TNFα expression in OC precursors, it strongly inhibits the OC differentiation in vitro (67). It is explained by the TLR-mediated suppression of RANK and TRME-2 expression through down-regulating cell surface c-Fms, the receptor for M-CSF, in OC precursors (68). However, OC precursors from TLR-2- and TLR-4-deficient mice have impaired capacity to uptake MSU crystals and differentiate to OC, implicating the indirect enhancement of OC differentiation by MSU crystal in the perspective of TLRs (69).
CONCLUDING REMARKS
Studies published over the past decade have found extensive evidences on the control of OC differentiation by immune cells and their cytokines (Fig. 2). Bone erosion and joint damage can proceed even though there is no evidence of active joint inflammation with effective medication for RA, so called uncoupling between clinical synovitis and damage progression (670). Recently, the antibody for RANKL, denosumab, has been known to be effective for reducing bone erosion, while it failed to show any difference in almost all parameters of disease activity compared with the placebo group (71). Moreover, there is an emerging interest in the enhancement of systemic bone loss in inflammatory diseases (72). Given the close interplay between the immune cells and OCs in inflammatory milieu, it is considered ideal the treatment strategy with agents that not only target the inflammation but also suppress OC differentiation, and this approach could potentially have significant impact on the future direction of drug development (Table 1).
Table 1
Critical mechanism-related implications and future directions for a new research agenda in bone erosion of inflammatory arthritis

 XML Download
XML Download