

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Fabry disease is an X-linked recessive lysosomal storage disorder of glycosphingolipid catabolism caused by a deficit in the lysosomal enzyme α-galactosidase A (α-Gal A). This enzyme is encoded by the GLA gene in the X chromosomal region Xq22.123 This enzymatic defect leads to progressive accumulation of glycosphingolipids, predominantly globotriaosylceramide (GL-3), throughout the body and particularly in the blood vessels, kidney, and heart.2345 Men with typical Fabry disease show almost complete absence of α-Gal A activity. They exhibit angiokeratoma, acroparesthesias, hypohidrosis, and corneal opacities in childhood or adolescence. With increasing age, progressive lysosomal GL-3 accumulation, particularly in the vascular endothelium, leads to renal failure, heart and brain vascular disease, and premature demise. Enzyme replacement therapy is effective at reducing and clearing accumulated glycosphingolipids, with subsequent improvement of cardiac function. Therefore, early recognition of Fabry disease is clinically relevant.67
The prevalence of Fabry disease is estimated at 1 in 40,000 to 1 in 117,000 live births for men,38 although calculations based on screening studies estimate a large number of unrecognized patients.9 Because Fabry disease is not common and its early classical manifestations tend to be nonspecific, the disorder is often unrecognized, misdiagnosed, or diagnosed in later life.310 Epidemiology studies demonstrate that many cases of Fabry disease are identified in cohorts of patients with unexplained left ventricular hypertrophy (LVH), with overall prevalence rates of approximately 3% in men (up to 6% in those older than 40 years) and up to 12% in women.11 However, the prevalence of Fabry disease in patients presenting with unexplained LVH remains unclear.
The prevalence of Fabry disease in Korean patients with LVH has not been reported to date. The purpose of this study was to determine the prevalence of Fabry disease in a large, prospective cohort of Korean men with LVH.
METHODS
Study population
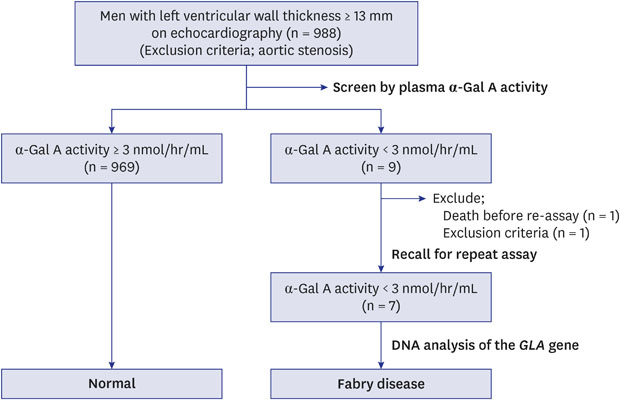
Ten clinical cardiology departments throughout Korea participated in this national, prospective, multicenter study from January 2004 to December 2008. A total of 988 consecutive, unselected Korean men with LVH on echocardiography at 10 centers were screened by measuring plasma α-Gal A activity. The criterion for LVH diagnosis was maximum left ventricular (LV) wall thickness ≥ 13 mm.12 Patients were defined as having LVH if diastolic thickness of the interventricular septum or LV posterior wall thickness was ≥ 13 mm on echocardiography. Exclusion criterion was aortic valve stenosis to more than a mild degree. The patient flow is summarized in Fig. 1.

Echocardiographic studies
Interventricular septal wall thickness, LV posterior wall thickness, left atrial dimension, LV end-diastolic dimension, and LV end-systolic dimension were evaluated by M-mode or two-dimensional echocardiography. Ejection fractions were obtained by modified biplane Simpson's method from apical 4- and 2-chamber views. Presence or absence and degree of abnormal LV wall motion were evaluated visually.13 Mitral inflow velocity was recorded with pulsed-wave Doppler sample volume positioned between the tips of the mitral leaflets. LV outflow velocity was recorded from the apical long axis view with pulsed-wave Doppler sample volume positioned just below the aortic valve. Deceleration time of mitral filling E velocity was measured from E wave peak to baseline intercept of the extrapolated descent of the wave Doppler time intervals were measured by mitral inflow and LV outflow velocity wave forms as described previously.14 Interval a from cessation to onset of mitral inflow was equal to the sum of isovolumic contraction time, ejection time, and isovolumic relaxation time. LV ejection time b was duration of the LV outflow velocity profile. Thus, the sum of the isovolumic contraction time and isovolumic relaxation time was obtained as a-b. Tei index, defined as the sum of isovolumic contraction and relaxation times divided by ejection time, was calculated as (a–b)/b.
Measurement of α-Gal A activity
Venous blood was collected from patients with LVH after echocardiographic studies. Plasma was prepared by cold centrifugation, stored at −80°C, and sent for analysis to Kagoshima University, Kagoshima, Japan. Plasma α-Gal A activity was measured with fluorogenic substrate 4-methylumbelliferyl-α-D-galactopyranoside (Sigma, St. Louis, MI, USA) using N-acetyl-D-galactosamine (Nacalai Tesque, Kyoto, Japan) as an inhibitor of α-N-acetylgalactosaminidase as described previously.15 Plasma α-Gal A activity < 3.0 nmol/hr/mL was defined as abnormally low. Patients with low α-Gal A activity had blood drawn again on another day for repeat measurement of α-Gal A activity. Patients with a low plasma α-Gal A activity on second measurement were evaluated for clinical manifestations of classic Fabry disease, including angiokeratoma, acroparesthesias, corneal and lenticular opacities, and hypohidrosis by clinical history, physical examination, and slit-lamp microscopy.
Genetic analysis
Patients with persistent low plasma α-Gal A activity (< 3.0 nmol/hr/mL) were recalled for further testing and DNA analysis of the GLA gene. Whole blood was collected and sent for analysis to the Medical Genetics Clinic and Laboratory, Asan Medical Center, Seoul, Korea. All patients who underwent genetic testing had previously provided informed consent, as required according to the Korean Law on Gene Technology.
Genomic DNA was isolated from peripheral blood leukocytes using a Puregene DNA isolation kit (Gentra, Minneapolis, MN, USA). Seven GLA exons and their intronic flanking sequences were amplified by polymerase chain reaction (PCR) using seven sets of previously described primers, followed by single-strand conformational polymorphism analysis and direct sequencing.16 DNA sequencing used the same primers as PCR with a BigDye Terminator V3.0 Cycle Sequencing Ready reaction kit (Applied Biosystems, Foster City, CA, USA). Electrophoresis and analysis of reactions was performed on an ABI 3100 Genetic analyzer (Applied Biosystems). If any variant of GLA gene was found in sequence analysis, we could explore in the documentations of the Human Gene Mutation Database (HGMD; http://www.hgmd.org), Clin Var (https://www.ncbi.nlm.nih.gov/clinvar/), and Genome Aggregation Database (gnomAD; http://gnomad.broadinstitute.org/).
Ethics statement
The Institutional Review Board (IRB) approval system was not available in every hospital at that time of our study enrollment (2004), so we did not receive an IRB approval, but all patients who participated in this study had previously provided informed consent for their participation.
RESULTS
α-Gal A activity and genetic analysis of patients
The study samples were 988 men with LVH on echocardiography. Two participants were excluded because one was a woman and one died before repeated assay. The final study population comprised 986 patients. Seven (0.7%) of the 986 patients had low plasma α-Gal A activity. Initial enzyme activities were 0.1 to 2.6 nmol/hr/mL with second measurements from 0.0 to 1.9 nmol/hr/mL. Plasma α-Gal A activities in the remaining 969 patients were 3.0 to 60.7 nmol/hr/mL (mean ± 1 standarad deviation [SD], 9.4 ± 4.8 nmol/hr/mL) (Fig. 2).
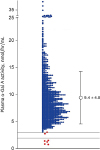 | Fig. 2Plasma α-Gal A activity in 986 men with LVH on echocardiography. In seven with LVH (red), values were 0.1 to 2.6 nmol/hr/mL. In the remaining 979 patients with LVH (black), values were 3.0 to 60.7 nmol/hr/mL (mean, 9.4 ± 4.8).α-Gal A = α-galactosidase A, LVH = left ventricular hypertrophy.
|
Results of genetic analysis of the seven patients are in Table 1. In three patients with low α-Gal A activity (patients 2, 4 and 5), we found previously described mutations; Gly328Arg, Arg301Gln, and His46Arg. No patients had previous diagnosis of Fabry disease.
Table 1
Summary of mutation analysis in patients with consistently low α-Gal A activity

![]()
Two with borderline low α-Gal A activity (patients 6 and 7) harbored a G-to-C transition at position 196 in exon 2, leading to an amino-acid substitution from glutamic acid to glutamine at position 66 (E66Q). Because E66Q mutation is reported as a functional polymorphism, we did not include the two patients with this mutation when evaluating for prevalence of Fabry disease.
In two patients with low plasma enzymatic activity (patients 1 and 3), no sequence variant was found in the GLA gene. We also did not include these two patients when evaluating for prevalence of Fabry disease.
Clinical and biological characteristics of patients with consistently low α-Gal A activity
Clinical characteristics and echocardiographic features of the seven patients with abnormally low enzymatic activity are in Tables 2 and 3. Patients ranged in age from 24 to 70 years. None had a history of cerebrovascular disease. Three patients had systemic hypertension and two had renal insufficiency. All had LVH with wall thickness from 13 to 23 mm. LVH was concentric in four patients and asymmetric in three.
Table 2
Characteristics of seven patients with low α-galactosidase A activity

A plus sign indicates the presence of a finding, and a minus sign its absence.
BUN = blood urea nitrogen.
![]()
Table 3
Echocardiographic features in patients with low α-galactosidase A activity

IVS = interventricular septal wall thickness, PW = left ventricular posterior wall thickness, LVEDd = left ventricular end-diastolic dimension, LVESd = left end-systolic dimension, LAD = left atrial dimension, FS = fractional shortening, EF = ejection fraction, E/A = the early (E) to late (A) ventricular filling velocities, DcT = deceleration time of mitral filling E velocity.
![]()
Clinical characteristics and family study of Fabry disease patients
Patient 2 was 24 years old and visited the hospital to evaluate proteinuria. Echocardiogrpahy showed LV septal wall thickness was 13.2 mm. α-Gal A activity was 0.1 nmol/hr/mL. Renal biopsy showed consistent with Fabry disease (Fig. 3). Genetic analysis revealed Gly328Arg mutation. This patient received enzyme replacement therapy and had been stable condition since then. Family history revealed that his mother carried Fabry disease but had no symptoms and did not undergo therapy. The patient had no siblings and further screening was not done.
 | Fig. 3Renal pathology in Fabry disease. (A) Segmental increase of cells and matrix with fuchsinophilic deposits and hyaline globules and markedly enlarged and vacuolated epithelial cells which contain abundant foamy cytoplasm (Toluidine blue stain, magnification, ×80). (B) Immunoglobulin M in the mesangium which is suggestive of a non-specific trapping and tends to exclude an immune-mediated GN (Masson Trichrome stain, magnification, ×400). (C) Glomerulopodocyte containing abundant electron dense myelin figures, Mesangialinsudative deposit with curvilinear microtubular and myelin figures, diffuse foot process effacement and myelin bodies and lysosomes in endothelial cells, smooth muscle cells, tubules and interstitial cells (Electron microscopy, magnification, ×2,500).
|
Patient 4 was 44 years old and had no symptoms related to Fabry disease on the initial visit but LVH was noted incidentally on echocardiography. The patient was confirmed to have Fabry disease based on an Arg301Gln missense mutation revealed on genotype analysis. His older brother had chronic renal insufficiency before screening and was also confirmed as having Fabry disease by genotype analysis. These two patients received enzyme replacement therapy.
Patient 5 had a history of chronic renal failure diagnosed at age 33 followed by kidney transplantation two years later. He also had long-standing hypohidrosis. Genotype analysis detected a His46Arg missense mutation in the GLA gene and the patient was subsequently diagnosed with Fabry disease. Family screening showed no genetic abnormalities in his brothers. His parents died before diagnosis and no further screening was done.
Clinical characteristics of no GLA mutation patients
One patient (patient 3) with no mutation at GLA gene visited clinic due to dyspnea on exertion. Echocardiography showed LVH, and proteinuria was not detected and renal biopsy was normal. Coronary angiography was unremarkable. He was discharged without problem at that time.
One patient (patient 1) with no mutation died due to recurrent heart failure aggravation. Also two patients has possibility of the candidates of Fabry disease, we should know that further investigation and observation need to be done.
DISCUSSION
Screening based on genotyping patients with low plasma enzyme activity revealed the following findings: 1) Three patients had a previously identified mutation; Gly328Arg, Arg301Gln, His46Arg; 2) Two unrelated men had the E66Q variant associated with functional polymorphism; 3) Two patients had low plasma enzyme activity, but no sequence variant was found in their GLA genes. Thus, the prevalence of Fabry disease was 0.3% in our cohort of 986 men patients with LVH ≥ 13 mm on echocardiography.
LVH is a key feature of Fabry disease and is reported in up to 50% of men and one-third of women.17 The prevalence of Fabry disease in patients with unexplained LVH has been investigated (Table 4). Screening for Fabry disease should be conducted by biochemistry analysis demonstrating deficiency or absence of α-Gal A determined in plasma, leukocyte, dried blood spot (DBS) or serum.29 α-Gal A activity in heterozygous women was usually normal and little low compared to that in men, so women were examined directly by genetic analysis without α-Gal A activity measurement to diagnose Fabry disease. α-Gal A activity measurement by screening method is only useful to men, so we confined to men with LVH in this study. Currently, seven studies using plasma α-Gal A activity as the primary screening method have reported the prevalence of Fabry disease in patients with cardiac hypertrophy.11151823262728 Nakao et al.15 identified seven patients with Fabry disease among a cohort of 230 men with LVH ≥ 13 mm on echocardiography (3%). Pathogenic mutations were identified in two, and the remainder had markedly reduced GLA mRNA levels. In a second study, Sachdev et al.18 found six patients (4%) with Fabry disease among 153 men with hypertrophic cardiomyopathy (HCM), with a prevalence of 6% in men diagnosed at older than 40 years. A study followed a cohort of 508 patients (328 men) with HCM at three HCM referral centers in Spain by screening plasma α-Gal A activity. In fifteen patents (3%), low plasma α-Gal A activity levels were demonstrated. Subsequent genetic analysis found that only three men (0.9%) and two women (1.1%) had pathogenic GLA mutations.23 A different screening strategy based on DBS testing was used in at French primary cardiology centers to screen 392 adult patents (278 men) with HCM defined by LV wall thickness ≥ 15 mm. Nine men and no women exhibited positive DBS tests. In four men (1.5% of the tested cohort), Fabry disease diagnosis was confirmed by standard blood testing, but no systemic genetic sequencing was performed.11 Screening of α-Gal A activity in the serum of 738 adult patients with LVH ≥ 13 mm on echocardiography from 25 centers in southwest Japan (Kyushu Island) found low α-Gal A activity in three patients (0.4%). Subsequent genetic analysis demonstrated all three had the mutation E66Q.26 A cohort of 560 patients (362 men) with HCM was followed at 10 centers in Belgium. In this study, α-Gal A activity was measured using DBSs and diagnosis was confirmed by mutational analysis of the GLA gene in men with mutation analysis as the primary screening tool in women. Two men (0.6%) and three women (1.7%) had pathogenic GLA mutations.27 In contrast, a small cohort study found four patients with Fabry disease among 100 men (4%) with LVH.28 Patients in these studies were selected because of LVH on echocardiography,1522 as diagnosis of HCM,19202123 or unexplained LVH.18 The divergence in reported prevalence rates may be related to small patient populations and/or differences in wall thickness of screened patients. Previous studies with fewer patients and single center trials found higher prevalence (3%–4%) for Fabry disease. However, studies with larger numbers of patients and multicenter trials found somewhat lower prevalence of Fabry disease ranging between 0% and 1.4%.
Table 4
Summary of previous studies examining the prevalence of Fabry disease in patients with LVH

| Author | Year | Study design | Country | LVH selection criteria | Sample size | Screening method | Confirmation method | Prevalence of FD No (%) | Comments | |
|---|---|---|---|---|---|---|---|---|---|---|
| Men | Women | |||||||||
| Nakao et al.15 | 1995 | P, S | Japan | LVH ≥ 13 mm | 230 | Plasma α-Gal A activity | Genetic study & mRNA levels | 7/230 (3) | In 5/7 cases, no mutation was found in the coding regions of the GLA gene | |
| Sachdev et al.18 | 2002 | P, S | United Kingdom | HCM ≥ 13 mm | 153 | Plasma α-Gal A activity | Genetic study | 6/153 (3.9) | 79 men ≥ 40 yr: 5/79 (6.3%) | |
| 74 men < 40 yr: 1/74 (1.4%) | ||||||||||
| Ommen et al.19 | 2003 | R, S | USA | Symptomatic HOCM | 100 | Myectomy tissue | 0/44 (0) | 0/56 (0) | ||
| Chimenti et al.20 | 2004 | P, S | Italy | HCM ≥ 13 mm | 96 | Endomyocardial biopsy & leukocyte α-Gal A activity | 2/62 (3.2) | 4/34 (11.8) | Biopsy study: selection bias? | |
| Arad et al.21 | 2005 | P, M | USA | HCM ≥ 13 mm | 75 | Genetic study | 0/45 (0) | 0/30 (0) | Inclusion of very young (> 12 yr) | |
| Morita et al.22 | 2006 | P, S | USA | HCM > 13 mm | 50 | Genetic study | 1/41 (2.4) | 0/9 (0) | ||
| Moserrat et al.23 | 2007 | P, M | Spain | HCM ≥ 13 mm | 508 | Plasma α-Gal A activity | Genetic study | 3/328 (0.9) | 2/180 (1.1) | Use of plasma α-Gal A activity in women |
| GLA variant: D313Y variant, 3 men | ||||||||||
| Havndrup et al.24 | 2010 | P, S | Denmark | HCM ≥ 13 mm | 90 | Genetic study | 1/56 (1.8) | 2/34 (5.9) | ||
| Elliott et al.25 | 2011 | P, M | Europe | LVH ≥ 15 mm | 1,386 | Genetic study | 3/885 (0.3) | 4/501 (0.8) | Age limitation (≥ 35 yr for men and ≥ 40 yr for women) | |
| High cut-off for LVH (15 mm instead of 13 mm) | ||||||||||
| Hagège et al.11 | 2011 | P, M | France | LVH ≥ 15 mm | 392 | DBSS | Men: Leukocyte α-Gal A activity | 4/278 (1.4) | 0/114 (0) | In men, genetic analysis was not done |
| Women: Genetic study | In women, use of plasma α-Gal A activity | |||||||||
| High cut-off for LVH (15 mm instead of 13 mm) | ||||||||||
| Mawatari et al.26 | 2013 | P, M | Japan | LVH > 13 mm | 738 | Serum α-Gal A activity | Genetic study | 0/738 (0) | GLA variant: E66Q, 3 men | |
| Terryn et al.27 | 2013 | P, M | Belgium | LVH > 13 mm | 560 | DBSS | Genetic study | 2/362 (0.6) | 3/178 (1.7) | GLA variant: c.639 + 6A > C, 1 woman |
| Palecek et al.28 | 2014 | P, M | Czech | HCM ≥ 13 mm | 100 | DBSS or Plasma α-Gal A activity | Leukocyte α-Gal A activity & genetic study | 4/100 (4) | ||
LVH = left ventricular hypertrophy, P = prospective cohort, S = single center study, R = retrospective study, α-Gal A = α-galactosidase A, HCM = hypertrophic cardiomyopathy, HOCM = hypertrophic obstructive cardiomyopathy, M = multicenter study, DBSS = dry blood spot screening.
![]()
In our study, Fabry disease was found in 0.3% of men with LVH ≥ 13 mm on echocardiography. This study included all patients with maximum LV wall thickness 13 mm or greater excluding aortic valve stenosis. We did not consider clinical manifestations. Thus, our study participants included patients with mild LVH, HCM, apical HCM, and basal septal hypertrophy as well as unexplained LVH. Compared to our study, other studies included patients with unexplained LVH and typical HCM. The probable reason for the low prevalence in this study was this difference. Our study has the limitation that detailed clinical manifestations were not considered at initial screen, but our study was a domestic analysis investigating the prevalence of Fabry disease in LVH patients.
In the 1990s, atypical cardiac Fabry disease with clinical symptoms appearing only in the heart was reported. Patients with atypical Fabry disease have lower α-Gal A activity and have LVH because of GL-3 accumulation in myocytes. Two patients with low α-Gal A activity and no gene mutations and two with the E66Q mutation in this study would have been classified as having atypical Fabry disease in previous studies. Recent studies do not recommend enzyme replacement therapy if confirmable findings are not noted on cardiac biopsy. As observed in our study, genetic studies or biopsy are required to determine whether patients with low α-Gal A activity have Fabry disease.
More than 500 disease-causing mutations have been identified in the gene encoding α-GAL, along with several functional polymorphisms. Some of these were previously believed to be pathogenic, but are now recognized to be functional polymorphisms.2630 The most noticeable example is the E66Q mutation (c.196G>C), a previously identified variant form of Fabry disease with a very high prevalence rate in Japanese and Korean populations (0.5%–1%). This polymorphism is proven to be functional, not pathogenic.31 In our study, two of 988 patients with HCM (0.2%) had the E66Q mutation. Therefore, we did not include these patients when evaluating for prevalence of Fabry disease.
In this study, α-Gal A activity was low, although the GLA mutation was not found in 2 patients. In other studies, patients with similar cases are regarded as having Fabry disease.11 Our study did not include these patients when determining Fabry disease prevalence. Nevertheless, since the patients had confirmed low α-Gal A activity, they could not be excluded as candidates for Fabry disease. Further evaluation will be required.
In this study, we diagnosed three new patients with Fabry disease and administered enzyme replacement therapy to four patients found by family screening. Although disputes about the efficacy of enzyme replacement therapy exist, many studies have had better outcomes when patients are treated before extensive tissue fibrosisdevelops.17 Thisenzyme replacement therapy might improve the disease prognosis of four patients.
Through screening based on genotyping patients with low plasma enzyme activity, the prevalence of Fabry disease in a large, consecutive cohort of Korean men with unexplained LVH was found to be 0.3%. The prevalence of Fabry disease was low in our study. However, early treatment can result in a good prognosis, therefore, differential diagnosis of Fabry disease should be considered in men with unexplained LVH.
 XML Download
XML Download